
民猪肠道菌群特征分析
2018-03-20 07:12张冬杰张跃灵王文涛
中国畜牧杂志 2018年3期
张冬杰,张跃灵,王文涛,汪 亮,刘 娣
(黑龙江省农业科学院畜牧研究所,农业部种养结合重点实验室,黑龙江哈尔滨 150086)
通常情况下,由机体自身基因编码、发挥消化功能的酶类消化能力均较弱,基本局限于对淀粉、蔗糖和乳糖的消化[1-2]。附植于消化道的微生物群落不是由宿主基因组编码,但对机体日常饮食中大量复杂碳水化合物的消化起主要作用。人和动物的消化道是由数以万亿的微生物细胞组成的多样化生态系统[3-4]。二代高通量测序技术不再需要培养肠道微生物,即可对菌群进行分类研究,因此,人类和各种动物的肠道微生物菌群才得以深入分析。另外,生物信息学的飞速发展也为探索肠道微生物的分类及功能注释提供了理论依据。目前,对肠道微生物的研究主要集中在肠道微生物与人类健康和疾病关系方面[5],对猪、牛、羊、禽、小鼠等其他哺乳动物的肠道微生物也开展了广泛研究,采用方法多是采集新鲜粪样,通过对16S rRNA变异区的测定分析菌群多样性[6-8]。
民猪是黑龙江省唯一一个被列入国家畜禽遗传资源保护名录的地方猪种,也是华北型猪种的一个典型代表。民猪具有耐粗饲、抗逆、繁殖力高、肉质优良等优点,但生长速度慢。目前即使饲喂相同饲粮,地方猪和引进猪种在饲料利用率、生长速度、肉品质等方面依旧存在着显著的不同。可见,地方猪已经形成了一种固化的、不同于引进猪种的消化吸收系统。本研究拟通过对不同月龄民猪肠道微生物的比较分析,揭示民猪肠道微生物群落特征;通过在饲粮中添加粗纤维,分析粗纤维对民猪肠道微生物的影响,为后续深入研究民猪的耐粗饲特性提供理论依据。
1 材料与方法
1.1 试验材料 试验用猪均由黑龙江省农业科学院畜牧研究所民猪保种场提供。试验共分3个处理组,成年组为5头10月龄体重为(100±7.24)kg的民猪(M1~M5,成年母猪),青年组为5头6月龄体重为(50±4.28)kg的民猪(MX1~MX5,青年母猪),青年粗纤维组为4头6月龄体重为(50±4.34)kg的民猪(MX7~MX10,青年母猪)。成年组与青年组饲喂基础日粮;青年粗纤维组在日粮中添加微贮玉米秸秆发酵物(粗纤维日粮),其他营养水平与基础日粮相近。试验饲粮组成及营养成分见表1。饲喂30 d后,在同一时间点分别取100 g左右的新鲜粪样,带回实验室,立即使用粪便基因组提取试剂盒(天根生化科技(北京)有限公司,DP328)进行基因组提取,1%琼脂糖凝胶电泳检测抽提基因组DNA,合格样品-20℃保存备用。
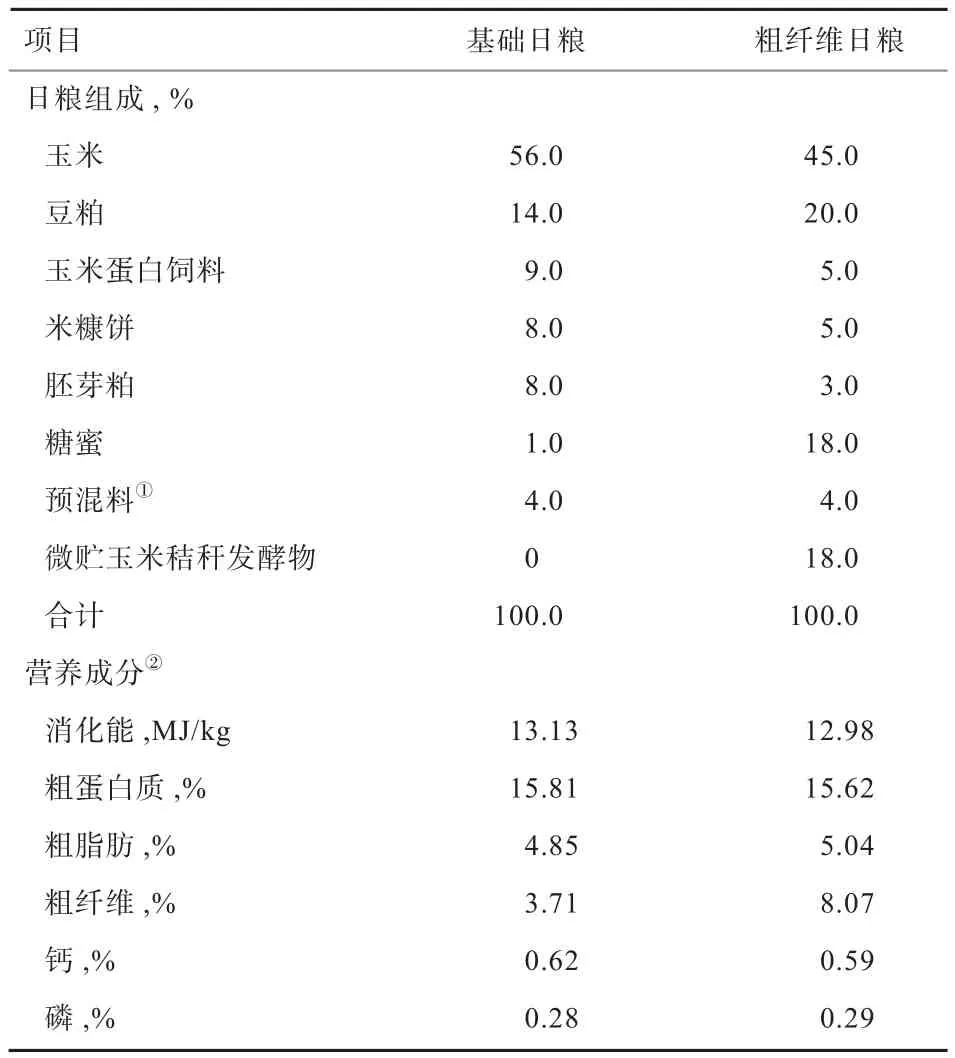
表1 日粮组成及营养成分
1.2 16SrRNA V4区的扩增 选择16S rRNA基因的V4区作为扩增和测序的目的区间。首先合成两端带有标签的特异引物,扩增16S rRNA的338~806 bp区间。引物序列:338F:5'-ACTCCTACGGGAGGCAGCAG-3',806R:5'-GGACTACHVGGGTWTCTAAT-3'。 使 用TransStartFastpfu DNA 聚合酶和ABI公司的GeneAmp®9700型PCR仪进行扩增反应。反应体系:5×FastPfu Buffer 4 μL,2.5 mmol/LdNTPs 2 μL,Forward Primer(5 μmol/L)0.8 μL,Reverse Primer(5 μmol/L)0.8 μL,FastPfu Polymerase 0.4 μL,模板 DNA 10 ng,补去离子水至20 μL。PCR反应条件:95℃3 min; 27个循环:95℃30 s,55℃30 s,72℃45 s;72℃10 min, 最 后10℃ 30 min。
1.3 样品回收与测序 每个样本3个重复,将同一样本的PCR产物混合后用2%琼脂糖凝胶电泳检测,使用AxyPrepDNA凝胶回收试剂盒(AXYGEN公司)回收PCR产物,Tris-HCl洗脱;2%琼脂糖电泳检测。然后将样本送交上海美吉生物有限公司进行测序,测序平台为IlluminaHiseq 2000。
1.4 数据优化 Illumina平台测序得到的是双末端序列数据,首先根据读序之间的相互重叠关系,将成对的读序拼接成1条序列,同时对每条读序的质量以及拼接效果进行质控过滤,根据序列首尾两端的条形码和引物序列区分样品,得到有效序列并校正序列方向。
1.5 OTU(Operational Taxonomic Units) 聚 类 分 析使用Usearch(Version 7.1)对质控后的序列进行OTU聚类分析。首先提取非重复序列,按照97%相似性对非重复序列(不含单序列)进行OTU聚类,在聚类过程中去除嵌合体,得到OTU的代表序列,最后将所有优化序列比对至OTU代表序列,选出与OTU代表序列相似性在97%以上的序列,生成OTU表格。
1.6 稀释性曲线的制备 对所测得的序列进行随机抽样,以抽到的序列数与它们所能代表OTU的数目构建稀释性曲线,当曲线趋向平坦时,说明测序数据量合理,更多的数据量只会产生少量新的OTU,反之则表明继续测序还可能产生较多新的OTU。本研究使用97%相似度的OTU,利用mothur做稀释性曲线,利用R语言工具制作曲线图。
1.7 基于Beta多样性距离的非度量多维尺度分析 非度量多维尺度法(NMDS)是一种将多维空间的研究对象(样本或变量)简化到低维空间进行定位、分析和归类,同时又保留对象间原始关系的数据分析方法。适用于无法获得研究对象间精确的相似性或相异性数据,仅能得到它们之间等级关系数据的情形。使用Qiime软件计算Beta多样性距离矩阵,Vegan软件包作NMDS分析和作图。
1.8 样本相似度分析 根据Beta多样性距离矩阵进行层次聚类(Hierarchical Cluatering)分析 ,使用非加权组平均法UPGMA(UnweightedPair Group Method with Arithmetic Mean)算法构建树状结构,得到各个样本间的树状关系图。
1.9 多样性指数及分类学分析 分别计算每个样本的Ace和Shannon多样性指数以及各样本的覆盖率。Ace(http://www.mothur.org/wiki/Ace)用来估计群落中OTU数目,是生态学中估计物种总数的常用指数之一;Shannon(http://www.mothur.org/wiki/Shannon)是用来估算样品中微生物多样性的指数之一,常用于反映Alpha多样性指数,其值越大,说明群落多样性越高。利用已有的16s细菌和古菌核糖体数据库Silva以及ITS真菌数据库Unite对获得的每个OTU对应的物种进行分类,采用RDP classifier贝叶斯算法(置信度阈值为0.7)对97%相似水平的OTU代表序列进行分类学分析,并在域、门、纲、目、科、属、种7个水平上统计每个样品的群落组成。选择Silva(Release 123 http://www.arb-silva.de)和 Unite(Release 7.0 http://unite.ut.ee/index.php)作为参考基因组数据库。
1.10 统计分析 使用SPSS16.0软件中的t检验对所获得的数据进行统计分析。
2 结果与分析
2.1 民猪肠道微生物DNA的提取及数据筛选 14个样本均获得了质量良好的基因组DNA,同时也扩增出了符合目的片段大小的PCR产物。测序完成后,对所测数据进行质控检查。数据经优化整理后,每个处理组的数据统计结果见表2。各组间所获得的序列条数及碱基个数经统计学检验均差异不显著(P>0.05)。

表2 样本测序数据统计结果
97%相似度下样品取样深度的稀释曲线见图1,当样品随机抽取的数据量为18 000左右时,曲线开始趋于平坦,表明取样深度基本一致。3个处理组测序列长度均集中在441~460 bp(占69.97%)和421~440 bp(30.01%),每个处理组的平均长度同为440 bp。
2.2 民猪肠道微生物的OTU分析 对3个试验组共计14个样本分别进行OTU统计分析,结果发现,M1~M5个 体分 别 获 得 764、762、675、773、800个 OTU,MX1~MX5个体分别获得 756、711、840、767、732个 OTU,MX7~MX10个体分别获得843、660、665、626个OTU。3个试验组间共享766个OTU,成年组专有33个OTU,青年组专有38个OTU,青年粗纤维组专有55个OTU。由此可见,随着日龄的增加,OTU的数量略有减少,但在饲粮中添加粗纤维则会显著增加OTU的数量。

图1 稀释曲线

图2 NMDS分析结果
2.3 民猪肠道微生物NMDS分析及聚类结果 对3个试验组共计14个样本进行NMDS及聚类分析,由图2可知,各组内个体间的微生物群落组成相似(组内个体的位置相对靠近);同一组个体均聚在同一个大的分支上(图3)。表明本试验中所采集的样品重复性较好,组间菌群差异明显。
2.3 民猪肠道微生物多样性指数分析 本研究结果表明,14个样本的覆盖率均在99%以上,说明测序质量良好,指数计算结果如表3所示。经统计学t检验分析后发现,3组间无论是物种总数还是微生物多样性在组间均无显著差异(P>0.05)。
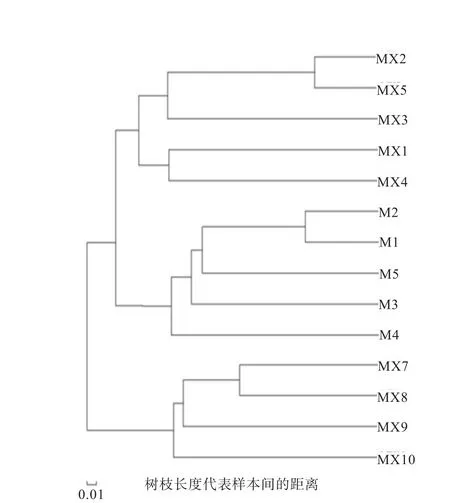
图3 各样本间相似度树状图
2.4 民猪肠道微生物不同分类学水平上的群落组成 本研究检测到的肠道微生物共涉及1个域(细菌),25个门,47个纲,79个目,121个科,207个属和390个种(表4)。
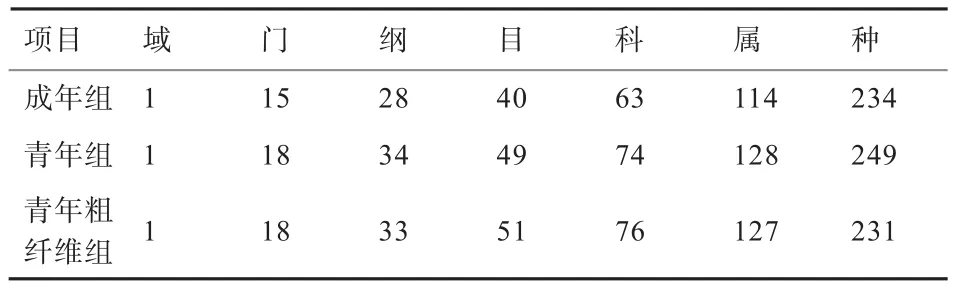
表4 14个样本在不同分类学水平上的群落组成
2.5 不同组间肠道微生物在门水平上的差异 本研究中共检测到25个门,其中拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)、螺旋菌门(Spirochaetae)、变形菌门(Proteobacteria)、软壁菌门(Tenericutes)、黏胶球星菌门(Lentisphaerae)、蓝菌门(Cyanobacteria)、纤维杆菌门(Fibrobacteres)等均占有一定的比例,但其中拟杆菌门、厚壁菌门和螺旋菌门所占的比例最高,总计可达到91%~92%(图4)。成年组与青年组个体的肠道微生物分布存在不同,而粗纤维的添加明显改变了青年个体肠道微生物的组成情况,大幅提升了拟杆菌门所占的比例,降低了厚壁菌门和螺旋菌门所占的比例,使其菌群分布比例更接近成年个体。

表3 各样本多样性指数统计表

图4 肠道微生物在门水平上的分布情况

图5 肠道微生物在属水平上的分布情况
2.5 不同组间肠道微生物在属水平上的差异 由图5可知,成年个体与青年个体均是密螺旋体属(Treponema)、疣微菌科(Ruminococcaceae_uncultured)和普氏菌属(Prevotella)占有较大优势,成年个体为10.58%、10.27%和10.85%,青年个体为14.60%、12.82%和7.39%。除此之外,成年个体有一个占到总数12.12%的S24-7_norank,是其肠道微生物内含量最高的菌属,远高于青年个体的3.75%,但此菌属目前尚无准确分类信息。可促进纤维消化的纤维杆菌属在成年个体肠道微生物内占1.17%,而在青年个体内仅为0.12%。
此外,饲粮中添加粗纤维会改变民猪的肠道微生物,与青年组相比,密螺旋体属和疣微菌属均显著下降,普氏菌属所占比例略有升高。在成年个体肠道内高表达、目前还没有准确分类信息的S24-7_norank,在粗纤维添加组内所占比例显著升高,达到21.63%,是该组个体肠道微生物内所占比例最高的菌属,远远超过青年组(3.75%)。据此推测这一菌属可能与粗纤维消化相关。粗纤维的添加也造成了肠道微生物内纤维杆菌属的增加,但增加幅度不大,达到0.29%,高于青年组(0.12%),仍低于成年组(1.17%)。
3 讨 论
3.1 不同月龄民猪肠道微生物多样性的差异 肠道微生物和疾病健康水平存在密切关系,其中菌群的多样性是反映宿主健康水平的关键指标。肠道微生物多样性的改变通常与饮食结构的调整以及健康状况的改变有关,如肥胖[9]、炎症性肠炎[10]和肠易激综合症等[11]。本研究结果发现,青年组和成年组间在肠道微生物碱基数量和序列条数方面没有差异,但青年组个体经质控过滤后所获得的有效序列数要比成年组多,说明随着月龄的增加,肠道微生物的多样性有下降趋势。进一步分析显示,Ace值在2组间基本一致,而青年组的Shannon值略高于成年组,但未达显著水平,这说明随着月龄的增加,民猪肠道微生物的多样性的确有下降趋势。同人类一样,新生婴儿、成年人和老年人的肠道微生物多样性均存在显著差异,但成年人会在很长一段时间内维持肠道微生物的稳定[12]。据此推测,本研究中青年组与成年组间无显著差异可能与民猪性成熟较早有关,50 kg的民猪已基本达到性成熟。
3.2 粗纤维对民猪肠道微生物多样性的影响 影响肠道微生物多样性的因素包括饮食,尤其是饮食成分中的纤维素。宿主没有消化纤维素的酶,但可以被细菌代谢,进而被小肠吸收。纤维素被认为是维持肠道微生物多样性的重要因素,而且能跨代产生作用。本研究结果显示,粗纤维添加组个体经质控过滤后所获得的有效序列数比青年组少,但未达到统计学上的显著水平。用来估计群落中OTU数目的Ace值,也是粗纤维组少于青年组,但用来估算样品中微生物多样性指数的Shannon值在2个组间基本一致。说明粗纤维的添加使民猪肠道微生物多样性出现了下降趋势。这与在小鼠上的研究结果不尽相同[14]。
3.3 不同月龄民猪肠道微生物群结构和物种丰度的差异拟杆菌门、厚壁菌门和螺旋菌门在成年组和青年组内均占到总数的91%~92%,从高到低的顺序也一致,但各自所占的比例不尽相同。紧随其后的是变形菌门和黏胶球形菌门,占比均超过1%。纤维杆菌门在成年民猪组内占比达到1.17%,而青年组为0.12%,这是2个组间在微生物群结构方面明显的不同之处。无壁菌门、蓝藻菌门和其他一些门类的占比均小于1%,这种微生物群结构与大白猪[15]相比更丰富。
在属的水平上,成年组的疣微菌属(Ruminococcaceae)物种丰度最高,达到112,其次是毛螺菌属(Lachnospiraceae)、密螺旋体属(Treponema)和普氏菌属(Prevotella),分别为36、30、29,这4个菌属可通过发酵饲料中不好消化的多糖和胶质生产短链脂肪酸,短链脂肪酸可以调节宿主的能量平衡,使宿主免于炎症反应,抑制脂肪组织发育[16]。青年组的主要物种丰度与成年组的基本一致,无显著差异。
3.4 粗纤维对民猪肠道微生物群结构和物种丰度的影响粗纤维的添加使青年个体肠道内拟杆菌门所占的比例由40.01%显著上升到57.13%,厚壁菌门和螺旋菌门所占的比例均出现了显著下降,肠道微生物群结构变得与成年组更为相似。拟杆菌门和厚壁菌门都是与肥胖有关的细菌,胖猪的拟杆菌门会显著低于瘦猪,而厚壁菌门会略高于瘦猪[17];同时也发现生活在北方的人类其机体肠道中也会存在较多的厚壁菌门[18]。由此可见,粗纤维的添加使青年个体出现了变瘦的趋势。在属的水平上,青年粗纤维组Turicibacter(芽孢杆菌目下的一个属,无准确命名)物种丰度最高,达到43,其次是Alistipes(理研菌科下的一个属,无准确命名),黄色单胞菌属(Xanthomonas)和黏胶球形菌属(Lentisphaeria),分别为33、29和27。可见,粗纤维的添加不仅改变了微生物群结构,也改变了优势菌种及其丰度。
4 结 论
本研究结果表明,民猪肠道内菌群以拟杆菌门、厚壁菌门和螺旋菌门为主,随着月龄的增加,各种菌群所占的比例会有所改变,但始终以这3种菌群为主。饲粮中粗纤维成分的添加可以改变民猪肠道微生物群结构,使青年个体的菌群分布特征与成年个体更为相近。青年个体肠道微生物内纤维杆菌属的比例远低于成年个体,即使粗纤维的添加会促进该类菌属的小幅增加,但仍旧低于成年个体,推测青年个体对粗纤维的消化能力小于成年个体。
[1] Cantarel B L, Lombard V, Henrissat B. Complex carbohydrate utilization by the healthy human microbiome[J]. PLoS One,2012, 7(6): e28742.
[2] El Kaoutari A, Armougom F, Gordon J I,et al. The abundance and variety of carbohydrate-active enzymes in the human gut microbiota[J]. Nat Rev Microbiol, 2013, 11(7): 497-504.
[3] Qin J, Li R, Raes J,et al. A human gut microbial gene catalogue established by metagenomicsequencing[J]. Nature, 2010,464(7285): 59-65.
[4] Costello E K, Stagaman K, Dethlefsen L,et al. The application of ecological theory toward an understanding of the human microbiome[J]. Science, 2012, 336(6086): 1255-1262.
[5] Wang P, Wang Y, Lu L,et al. Alterations in intestinal microbiota relate to intestinal failure-associated liver disease and central line infections[J]. J Pediatr Surg, 2017, 52(8): 1318-1326.
[6] Hanning I, Diaz-Sanchez S. The functionality of the gastrointestinal microbiome in non-human animals[J].Microbiome, 2015, 3: 51.
[7] Almasaudi S, KaoutariA E, Drula E,et al. A Metagenomics investigation of carbohydrate-active enzymes along the gastrointestinal tract of saudisheep[J]. Front Microbiol, 2017, 8:666.
[8] 杨俊花, 赵志辉, 郭文博, 等. 应用Illumina-MiSeq高通量测序技术分析脱氧雪腐镰刀菌烯醇对小鼠肠道菌群的影响[J]. 动物营养学报, 2017, 29(1): 158-167.
[9] Turnbaugh PJ, Ley RE, Mahowald MA,et al. An obesityassociated gut microbiome with increased capacity for energy harvest[J]. Nature, 2006, 444(7122): 1027-1031.
[10] Frank DN, St Amand AL, Feldman RA,et al. Molecularphylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases[J]. Proc Natl Acad Sci USA, 2007, 104(34): 13780-13785.
[11] Kassinen A, Krogius-Kurikka L, Makivuokko H,et al. The fecal microbiota of irritable bowel syndrome patients differs significantly from that of healthy subjects[J]. Gastroenterology,2007, 133(1): 24-33.
[12] Claesson MJ, Cusack S, O'Sullivan O,et al. Composition,variability, and temporal stability of the intestinal microbiota of the elderly[J]. Proc Natl Acad Sci USA, 2011, 108 (Suppl 1):4586-4591.
[13] Sonnenburg ED, Smits SA, Tikhonov M,et al. Diet-induced extinctions in the gut microbiota compound over generations[J].Nature, 2016, 529(7585): 212-215.
[14] Zhao W, Wang Y, Liu S,et al. The dynamic distribution of porcine microbiota across different ages and gastrointestinal tract segments[J]. PLoS One, 2015, 10(2):e0117441.
[15] He M, Fang S, Huang X,et al. Evaluating the contribution of gut microbiota to the variation of porcine fatness with the cecum and fecal samples[J]. Front Microbiol, 2016, 7: 2108.
[16] 郭秀兰.猪肠道硬壁菌门和拟杆菌门数量的检测及其相对丰度与脂肪沉积的相关性研究[D]. 成都: 四川农业大学,2009: 1-2.
[17] Suzuki TA, Worobey M. Geographical variation of human gut microbial composition[J]. Biol Lett, 2014, 10(2): 1-12.
[18] Clarke SF, Murphy EF, O’Sullivan O,et al. Targeting the microbiota to address diet-induced obesity: a time dependent challenge[J]. PLoS One, 2013, 8(6): e65790.
猜你喜欢
中国农学通报(2022年14期)2022-06-01
中国生殖健康(2020年4期)2021-01-18
中国比较医学杂志(2020年4期)2020-05-26
中国现代中药(2019年5期)2019-07-03
科海故事博览·下旬刊(2019年6期)2019-04-16
中国沼气(2019年1期)2019-04-13
中国生殖健康(2018年4期)2018-11-06
吉林农业(2018年23期)2018-01-17
冰雪运动(2016年4期)2016-04-16
中国民族民间医药·下半月(2014年1期)2015-02-02
