
PPI网络比对用于植物乳杆菌的糖代谢研究
2018-03-19 02:44苗孟君丁彦蕊
计算机工程与应用 2018年6期
苗孟君,丁彦蕊
1.江南大学物联网工程学院,江苏无锡214122
2.江南大学数字媒体学院,江苏无锡214122
PPI网络比对用于植物乳杆菌的糖代谢研究
苗孟君1,丁彦蕊2
1.江南大学物联网工程学院,江苏无锡214122
2.江南大学数字媒体学院,江苏无锡214122
CNKI网络出版:2017-04-01,http://kns.cnki.net/kcms/detail/11.2127.TP.20170401.0853.034.html
1 引言
植物乳杆菌有助于调节人和动物的机体微生物菌群的平衡,增强机体的免疫系统,并且具有多种保健功能,其主要存在于人和动物胃、肠道以及肉类中。Kleerebezem[1]等人于2003年测序出植物乳杆菌WCFS1的完整基因序列,此后,其逐渐成为研究植物乳杆菌的模型菌株之一。而由Zhang[2]等人测序出的植物乳杆菌JDM1的基因序列与WCFS1高度相似(>90%),但由于JDM1长期生活在富营养的环境中,使得JDM1中缺失了一些糖转移和代谢基因。针对这一现象,Zhang[2]等人从基因序列的角度说明JDM1较WCFS1缺失了一些糖转移和代谢基因。然而,生物体内的细胞功能和生物过程是通过蛋白质之间的相互作用完成的,因此,只从WCFS1和JDM1的某段序列或者某个编码的蛋白质分析阐明两个菌株的糖代谢功能机制的差异是片面的。因此,构建WCFS1和JDM1的糖代谢功能模块的PPI网络[3],运用智能网络比对算法研究两种植物乳杆菌糖代谢功能模块的差异具有重要的意义。
生物分子网络比对是利用生物分子的相似性和生物分子网络的拓扑信息并结合图理论,对生物分子网络进行比对,是理解不同生物系统之间的相似性和差异性的一种有效方法。陈璟[4]等人通过比对产甲烷的常温古细菌和嗜热古细菌的代谢网络,发现了常温古细菌和嗜热古细菌的保守代谢途径,推测嗜热菌的耐热性可能与胞内酪氨酸有关。蛋白质-蛋白质相互作用网络的比对,可以有效地预测蛋白质-蛋白质相互作用、蛋白质功能,挖掘不同物种之间的保守区域,分析功能模块的差异性。Seah[5]等人提出网络比对算法DualAligner,通过区域到区域的蛋白质相互作用网络比对,发现了人类和酵母之间的高度保守区域。
近年来,PPI网络比对算法发展迅速,这促进了研究者们对蛋白质-蛋白质相互作用的探索,并且有利于蛋白质功能的预测和保守功能模块挖掘的研究。网络比对算法分为两种类型:局部网络比对和全局网络比对。局部网络比对是不明确的,因为一个网络中的节点可以与另一个网络中的多个节点相匹配。PathBLAST[6]是一种最早期的局部网络比对算法之一,其通过寻找蛋白质相互作用途径的高比对得分进行网络比对,Network-BLAST[7]是此算法的改进形式。在全局网络比对中,网络中的每一个节点都与另一个网络中的唯一一个节点相匹配。Berger[8]等人首次提出了PPI网络的全局比对算法,IsoRank,并进一步改进此算法,形成IsoRankN[9]和PISwap[10]。Kuchaiev等人完全利用网络的拓扑信息,提出算法:GRAAL[11]和MI-GRAAL[12],并且通过这些算法发现人类和酵母的网络拓扑结构非常相似。Milenkovic等人发展了这些算法,提出H-GRAAL[13]。HGA-2N[14]改进匈牙利贪心算法(HGA),并实现了图形处理单元的并行计算。MAGNA[15]基于遗传算法寻求最大化精度的全局网络比对。Evolutionary Graph Edit Distance Algorithm(GEDEVO)[16]则是一种利用图编辑距离作为优化模型以寻求最优比对的进化算法。
由于植物乳杆菌WCFS1和JDM1在糖代谢模块存在较大差异,为了更加精确地分析WCFS1和JDM1关于糖代谢模块的差异,本文通过分别构建植物乳杆菌WCFS1和JDM1的糖酵解模块,戊糖磷酸途径模块,柠檬酸循环模块的蛋白质-蛋白质相互作用(PPI)网络,采用网络比对算法GEDEVO,对构建的PPI网络进行比对,从系统水平上分析植物乳杆菌JDM1和WCFS1的糖代谢功能模块之间的差异,挖掘糖代谢功能模块的保守区域,寻找WCFS1和JDM1糖代谢功能模块的差异路径,预测蛋白质-蛋白质相互作用。
2 数据集和方法
2.1 数据来源
本文从KEGG数据库获得植物乳杆菌WCFS1(Lactobacillus plantarum WCFS1)和JDM1(Lactobacillus plantarum JDM1)的糖酵解模块(Glycolysis),戊糖磷酸途径模块(Pentose phosphate pathway),柠檬酸循环模块(Citrate cycle)的全部蛋白质数据,并结合STRING数据库,分别获得这三个模块的蛋白质-蛋白质相互作用数据。这三个模块的全部蛋白质的相互作用是根据STRING数据库中蛋白质-蛋白质相互作用数据可信度确定的[17-18],选择标准为可信度大于0.7[19]。
2.2 PPI网络的构建
根据从STRING数据库获得的蛋白质-蛋白质相互作用数据,分别构建WCFS1和JDM1的糖酵解功能模块,戊糖磷酸途径功能模块,柠檬酸循环功能模块的PPI网络。本文用图G=(V,E)表示蛋白质-蛋白质相互作用网络,其中,V代表蛋白质集合,E代表蛋白质-蛋白质之间的相互作用集合。具体信息如表1所示。

表1 WCFS1和JDM1的糖代谢模块的PPI网络数据
2.3 方法
生物网络比对结合图理论和生物信息,通过一定规则,使得网络中的节点相互对应。其中,PPI网络比对,利用网络比对算法,使得网络中的蛋白质相互对应,预测蛋白质-蛋白质相互作用、蛋白质功能,挖掘保守功能模块,分析功能模块之间的差异。本文选取GEDEVO算法分别对植物乳杆菌JDM1和WCFS1的糖酵解模块,戊糖磷酸途径模块,柠檬酸循环模块的PPI网络进行比对。GEDEVO是一种利用图编辑距离作为优化模型以寻求最优比对的进化算法,其受自然灵感启发,模仿规则“适者生存”,广泛用于解决许多NP难问题。GEDEVO基于进化算法思想,将一对PPI网络模型作为两个无向不加权图G1=(V1,E1)和G2=(V2,E2),并利用图编辑距离优化映射f使V1和V2中的节点一对一映射。定义G1和G2之间的图编辑距离为:
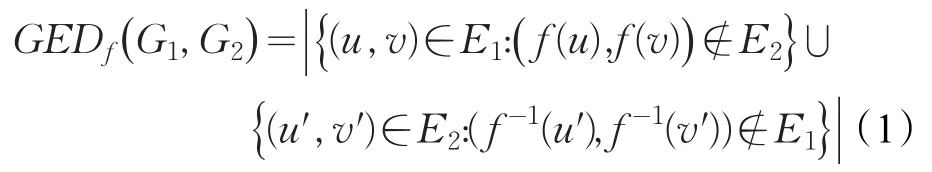
其中,GEDf()G1,G2表示图G1和G2的图编辑距离,(u,v)表示图G1中的节点u和v之间的边,E1为图G1中的边集;f()u和f(v)分别为节点u和v的映射节点,E2为图G2中的边集,(u′,v′)表示图G2中的节点u′和v′之间的边,f-1(u′)和f-1(v′)分别为节点u′和v′的映射节点。
GEDEVO算法主要分为3个步骤,分别为:
步骤1初始化种群和个体评价
初始化种群的个体是两个PPI网络的一个映射。
个体评价即评价一个映射的质量。设每对节点u∈V1和v∈V2之间的映射为v=f(u)。定义节点对得分pairScoref(u,v)表示节点u和v之间的映射质量,公式如下:
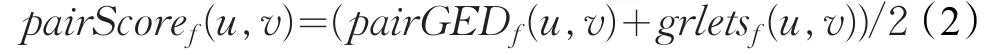
其中,PairGEDf(u,v)表示通过映射f,使节点u和v对应所要增加或删除的边的数量。grletsf(u,v)表示graphlet度标签距离(GSD)[20],grletsf(u,v)被定义为两个节点的邻近拓扑结构差异。
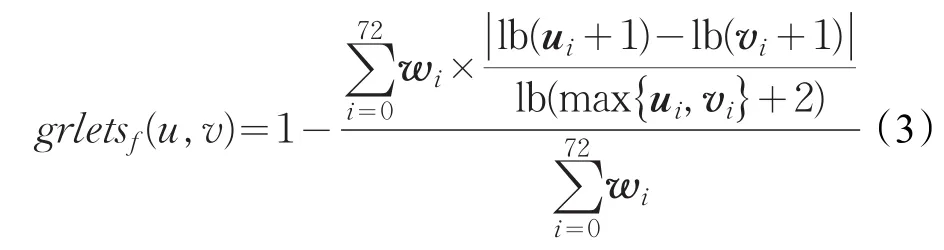
其中,ui表示节点u的第i个特征向量[20],vi表示节点v的第i个特征向量[20],wi表示同构图i的权重,grletsf(u,v)的值在0到1之间,grletsf(u,v)的值越大,说明节点u和v的邻近区域的拓扑特征越相似[21-22]。
步骤2生成子代
为了产生新的映射f,并且保持种群多样性避免局部最优,得到高且快速收敛的最佳方案,进行如下操作:
(1)将两个PPI网络中的蛋白质分别进行随机排列,并随机产生一个映射f。
(2)采取部分映射交叉(PMX)变异思想,把映射中所有蛋白质对得分的平均值作为阈值,把一个映射分成低得分组和高得分组。然后,随机交换高得分组。为了避免局部最小,也交换概率较低的低得分对(PPI网络的1%)。
(3)在一个映射f的交叉结果中,从父代中保护低得分对。
(4)对于定点变异,在一个映射f中,选择低得分对进行交换,最终保留高得分对。
步骤3终止迭代
GEDEVO的收敛时间主要取决于种群的大小以及输入的PPI网络的拓扑性质。GEDEVO可以设定收敛条件:(1)迭代次数;(2)运行时间;(3)个体的最佳映射没有随着迭代次数发生明显改变时(迭代次数为30时)。
2.4 比对质量评价
网络比对结果的质量标准常常反映在拓扑方面和生物方面。本文采用节点正确性(Node Correctness,NC)[15]和边正确性(Edge Correctness,EC)[16]评价网络比对结果。EC被定义为输入网络边的比对的百分比,用于衡量输入网络的拓扑结构的相似程度,EC值越高,输入的网络越相似。NC被定义为输入的第一个网络中的节点正确比对第二个网络节点的百分比。EC和NC的计算公式如下表示:
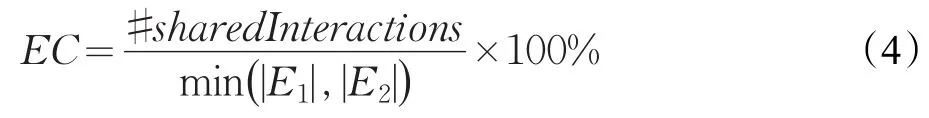
其中,#sharedInteractions表示网络G1和G2中对应的边的数目,|E1|和|E2|分别表示网络G1和G2中边的数目。
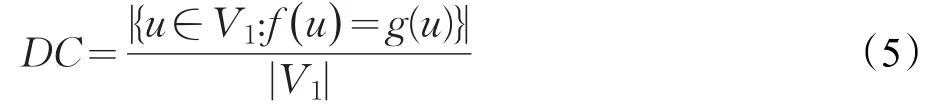
其中,g是正确的节点映射,f(u)=g(u)表示节点的比对结果与正确的节点映射相同。显然,正确比对节点的映射g要预先知道。这里,设置g为比对的两个网络中节点(蛋白质)的同源性。
3 结果与讨论
植物乳杆菌WCFS1和JDM1的基因序列非常相似(>90%),由于JDM1长期生活在富营养环境中,使得JDM1缺失了一些糖代谢基因,这导致WCFS1和JDM1在糖代谢功能模块存在一定差异。因此,本文利用网络比对方法对WCFS1和JDM1的糖代谢(糖酵解、戊糖磷酸途径、柠檬酸循环)功能模块的PPI网络进行比对,挖掘两者的糖代谢模块的保守路径,并分析两者的差异。
本文选择GEDEVO算法,对植物乳杆菌WCFS1和JDM1的糖酵解,戊糖磷酸途径,柠檬酸循环模块的PPI网络进行比对。并利用边正确性(公式(4))和节点正确性(公式(5))评价比对结果。其中,计算节点正确性时,以KEGG数据库中蛋白质的同源性数据为标准。另外,本文将GEDEVO算法的比对结果与GRAAL、MAGNA算法进行了比较。表2表示了GEDEVO算法、GRAAL算法、MAGNA算法在糖酵解、戊糖磷酸途径、柠檬酸循环模块中的边正确性结果。表3表示了GEDEVO算法、GRAAL算法、MAGNA算法在糖酵解、戊糖磷酸途径、柠檬酸循环模块中的节点正确性结果。
从表2的边正确性来看,GEDEVO算法和MAGNA算法明显优于GRAAL算法,尤其在糖酵解和戊糖磷酸途径的比对中优势明显。从表3的节点正确性来说,GEDEVO算法在所有网络的比对中都明显高于GRALL和MAGNA。综合边正确性和节点正确性来看,GEDEVO算法更适合于WCFS1和JDM1的网络比对。因此,本文采用GEDEVO算法进行网络比对。
从表2可以看出,GEDEVO算法对于WCFS1和JDM1的糖酵解、戊糖磷酸途径、柠檬酸循环模块的比对结果的边正确性都大于90%,分别是93.6%、96%、100%。这表明植物乳杆菌WCFS1和JDM1的这三个模块的PPI网络的拓扑结构非常相似。表3中,WCFS1和JDM1的糖酵解、戊糖磷酸途径、柠檬酸循环模块的比对结果的节点正确性都大于80%,分别是82.9%、88%、100%。WCFS1和JDM1的糖酵解、戊糖磷酸途径、柠檬酸循环模块中的蛋白质大部分互为同源蛋白质,但也存在一些差异。
此外,为了验证实验结果的可靠性,分别对三个功能模块做了10组(共30组)比对实验,分别计算每个功能模块实验结果的平均值和方差,具体数据如表4所示。
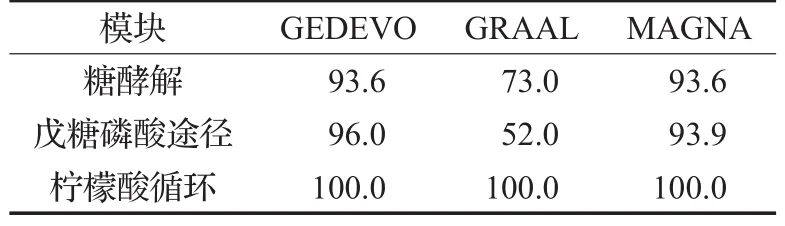
表2 GEDEVO、GRAAL、MAGNA算法对糖酵解、戊糖磷酸途径、柠檬酸循环模块的PPI网络比对的EC值分布%

表3 GEDEVO、GRAAL、MAGNA算法对糖酵解、戊糖磷酸途径、柠檬酸循环模块的PPI网络比对的DC值分布%

表4 对三个功能模块进行比对的EC值和DC值的平均值和方差
从表4中可以看出,糖酵解模块、戊糖磷酸途径模块和柠檬酸循环模块的DC值的平均值都大于0.85,EC值的平均值都大于0.9,方差都小于0.002 5,这表明实验结果非常可靠并且很稳定。
下面将根据比对结果,阐明WCFS1和JDM1的这三个功能模块的差异。
图1描述了植物乳杆菌WCFS1和JDM1的戊糖磷酸途径模块的PPI网络比对结果。图的左边表示JDM1的PPI网络,右边是WCFS1的PPI网络。从图中可以看出,JDM1中的蛋白质ribose-phosphate pyrophosphokinase(prs1)、ribokinase(rbsK3)、transketolase(tkt4)、fructosebisphosphate aldolase(fba)、2-keto-3-deoxy-6-phosphogluconate aldolase(JDM1_0578)、glucose-6-phosphate isomerase(pgi)、glucose-6-phosphate 1-dehydrogenase(gpd)、ribulose-phosphate 3-epimerase(rpe)之间通过相互作用连接成一条蛋白质相互作用路径prs1-rbsK3-tkt4-fba-JDM1_0578-pgi-gpd-rpe,与WCFS1中的蛋白质ribose-phosphate pyrophosphokinase(prs1)、Ribokinase(rbsK1)、transketolase(tkt4)、fructose-bisphosphate aldolase(fba)、glucose-6-phosphate isomerase(pgi)、glucose-6-phosphate 1-dehydrogenase(gpd)、ribulose-phosphate 3-epimerase(rpe)、2-keto-3-deoxygluconate kinase(kdgk)组成的路径prs1-rbsK1-tkt4-fba-pgi-gpd-rpe-kdgk非常相似,如两者的prs1-rbsK3(rbsK1)-tkt4-fba路径是一致的。但是这两条路径又存在差异,如图1右边网络中的蛋白质kdgk,JDM1中并没有与之对应的蛋白质。WCFS1中的蛋白质kdgk是一种磷酸化的激酶,它可以生成蛋白质2-keto-3-deoxy-6-phospho-gluconate aldolase(JDM1_0578)[23],并且,其在原核生物中的葡萄糖分解代谢和糖酸降解中起着关键作用[24],JDM1_0578则通过双功能KDPG醛缩酶裂解丙酮酸和甘油醛-3-磷酸(GAP)。此外,JDM1中没有蛋白质与WCFS1中的蛋白质deoxyribose-phosphate aldolase(deoC)相对应,而蛋白质deoC是一种可以从死亡细胞中产生脱氧核苷的参与分解代谢的酶。这种酶经常出现在生活在营养缺失的环境中的生物体中[25]。因为WCFS1生活在营养缺失的环境中,它会用这种酶从死亡的细胞中获得营养物质,使其生长。而JDM1则长期生活在富营养环境中,因此它不需要这种酶。
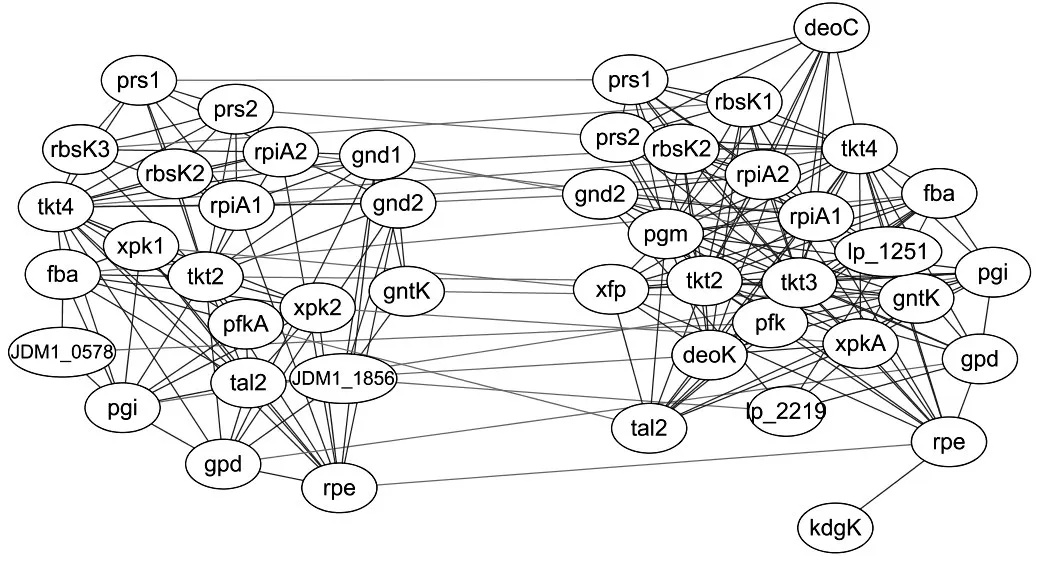
图1 WCFS1和JDM1的戊糖磷酸途径PPI网络比对图
图2描述了植物乳杆菌WCFS1和JDM1的糖酵解模块的PPI网络比对结果。图的左边表示JDM1的糖酵解PPI网络,右边是WCFS1的糖酵解PPI网络。从表2中可以看出,两者的拓扑结构非常相似,EC值达到了93.6%。如,两者中的蛋白质glucose-6-phosphate isomerase(pgi)都处于PPI网络的中心位置,与上下两边的许多蛋白质都具有相互作用,可见,蛋白质glucose-6-phos-phate isomerase(pgi)在WCFS1和JDM1的糖酵解模块中分别具有至关重要的作用。JDM1中的蛋白质glucokinase(glk)和WCFS1中的蛋白质bifunctional protein:transcription regulator和sugar kinase(lp_1573)都与醛糖异构酶(aldose 1-epimerase、galM1、galM2、galM3)相互作用,其中蛋白质glk和lp_1573互为同源蛋白质。但是,WCFS1和JDM1的糖酵解模块也存在差异。JDM1中的蛋白质bifunctional acetaldehyde-CoA/alcohol dehydrogenase(adhE)与蛋白质pyruvate dehydrogenase complex,E2 component(pdhC)相互作用,与之对应的,WCFS1中的蛋白质bifunctional protein:alcohol dehydrogenase、acetaldehyde dehydrogenase(adhE)和蛋白质pyruvate dehydrogenase complex、E2 component(pdhC)和phosphoenolpyruvate carboxykinase(ATP)(pck)相互作用。可以推测,JDM1中的adhE与蛋白质pck相互作用。JDM1中的蛋白质pdhC有4条边,与其有相互作用的蛋白质分别是pck、pyruvate dehydrogenase complex(pdhA)、pyruvate dehydrogenase complex、E1 component,beta subunit(pdhB)、dihydrolipoamide dehydrogenase(pdhD)。而WCFS1中的pdhC除了与这四个蛋白质相互合作,还与蛋白质pyruvate kinase(pyk)相互作用。可见,JDM1中的pdhC与蛋白质pyk具有相互作用[26]。通过网络比对,发现了WCFS1和JDM1的糖酵解模块中存在许多保守路径,并且预测了JDM1中的一些蛋白质之间的相互作用。

图2 WCFS1和JDM1的糖酵解PPI网络比对图
图3描述了植物乳杆菌WCFS1和JDM1的柠檬酸循环模块的PPI网络比对结果。图的左边是JDM1的柠檬酸循环PPI网络,右边是WCFS1的柠檬酸循环PPI网络。从图中可以看出,WCFS1和JDM1的柠檬酸循环模块中,对应蛋白质的邻居节点个数相同,并且由表2可知,其EC值为100%,这表明两者的拓扑结构非常相似。WCFS1和JDM1的柠檬酸循环模块的PPI网络都分为两部分,图中,上下两部分的蛋白质之间并没有相互作用。通常情况下,完成一个功能模块,需要蛋白质之间的相互合作,因此,能推测,JDM1和WCFS1的柠檬酸循环模块需要与柠檬酸循环模块之外的蛋白质互相合作,完成柠檬酸循环功能。此外,JDM1的柠檬酸循环模块中有11个蛋白质,WCFS1中有10个蛋白质,JDM1中的蛋白质fumarate reductase/succinate dehydrogenase(JDM1_0211)没有与WCFS1中的蛋白质对应。
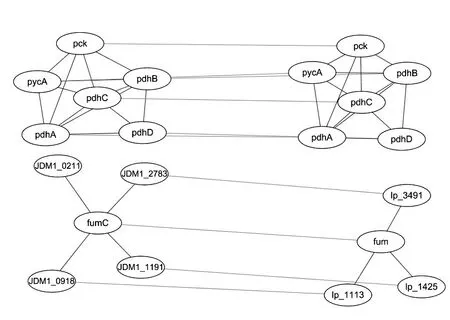
图3 WCFS1和JDM1的柠檬酸循环PPI网络比对图
4 总结
本文选择GEDEVO算法分别比对植物乳酸杆菌JDM1和WCFS1的糖酵解,戊糖磷酸途径,柠檬酸循环功能模块的蛋白质-蛋白质相互作用网络。发现JDM1和WCFS1的糖酵解、戊糖磷酸途径、柠檬酸循环的PPI网络的拓扑结构非常相似,并挖掘了许多保守路径。如,戊糖磷酸途径中,JDM1中的蛋白质glucokinase(glk)和WCFS1中的与其同源的蛋白质bifunctional protein:transcription regulator;sugar kinase(lp_1573)都与醛糖异构酶(aldose 1-epimerase,galM1,galM2,galM3)相互作用。但也存在差异,在糖酵解功能模块中,JDM1中的蛋白质相互作用路径prs1-rbsK3-tkt4-fba-JDM1_0578-pgi-gpd-rpe和WCFS1中的蛋白质相互作用路径prs1-rbsK1-tkt4-fba-pgi-gpd-rpe-kdgk非常相似,但也存在差异。其中蛋白质2-keto-3-deoxygluconate kinase(kdgK)的产物是2-keto-3-deoxy-6-phosphogluconate aldolase(JDM1_0578)。在本文中,利用PPI网络比对的方法,不仅挖掘了JDM1和WCFS1在糖代谢模块的保守路径,发现了糖代谢模块的差异,还预测了蛋白质-蛋白质相互作用,如,戊糖磷酸途径中,JDM1中的adhE与蛋白质pdhD发生相互作用。综上所述,JDM1和WCFS1的糖代谢功能模块的拓扑结构非常相似,但也存在差异。通过利用PPI网络比对方法,对两者的糖代谢功能模块进行比对,挖掘两者糖代谢功能模块的保守路径,以及存在差异的路径,并预测了蛋白质-蛋白质相互作用。通过实验进一步表明,PPI网络比对对于预测蛋白质-蛋白质之间的相互作用,挖掘保守模块,发现物种及功能模块之间的差异具有重要意义。
[1] Kleerebezem M,Boekhorst J,van Kranenburg R,et al.Complete genome sequence of Lactobacillus plantarum WCFS1[J].Proceeding of the National Academy of Sciences of the United States of America,2003,100(4):1990-1995.
[2] Zhang Zhuoyang,Liu Chang,Zhu Yongzhang,et al.Complete genome sequence of Lactobacillus plantarum JDM1[J].Journal of Bacteriology,2009,191(15):5020-5021.
[3] Barabasi A L,Oltvai Z N.Network biology:understanding the cell's functional organization[J].Nature Reviews Genetics,2004,5(2):101-113.
[4] 陈璟,须文波.产甲烷的常温古细菌和嗜热古细菌的代谢网络比对研究[J].小型微型计算机系统,2015,36(8):1869-1873.
[5] Seah B S,Bhowmick S S,Dewey Jr C F.DualAligner:A dual alignment-based strategy to align protein interaction networks[J].Bioinformatics Advance Access,2014,30(18):2619-2626.
[6] Kelley B P,Yuan Bingbing,Lewitter F,et al.PathBLAST:A tool for alignment of protein interaction networks[J].Nucleic acids research,2004,32:83-88.
[7] Kalaev M,Smoot M,Ideker T,et al.NetworkBLAST:Comparative analysis of protein networks[J].Bioinformatics,2008,24:594-596.
[8] Singh R,Xu J,Berger B.Pairwise global alignment of protein interaction networks by matching neighborhood topology[C]//International Conference on Research in Computational Molecular Biology,2007,4453:16-31.
[9] Liao C S,Lu Kanghao,Baym M,et al.IsoRankN:Spectral methods for global alignment of multiple protein networks[J].Bioinformatics,2009,25(12):253-258.
[10] Chindelevitch L,Liao C S,Berger B,et al.Local optimization for global alignment of protein interaction networks[C]//Pacific Symposium on Biocomputing,Hawaii,USA,2010:123-132.
[11] Kuchaiev O,Milenković T,Memišević V,et al.Topological network alignment uncovers biological function and phylogeny[J].Journal of the Royal Society Interface,2010,50(7):1341-1354.
[12] Kuchaiev O,Przˇulj N.Integrative network alignment reveals large regions of global network similarity in yeast and human[J].Bioinformatics,2011,27:1390-1396.
[13] Milenkovic T,Ng W L,Hayes W,et al.Optimal network alignment with graphlet degree vectors[J].Cancer Inform,2010,9:121-137.
[14] Xie J,Zhou Z,Ma J et al.Graphics processing unitbased alignment of protein interaction networks[J].IET Systems Biology,2015,9(4):120.
[15] Saraph V,Milenkovic T.MAGNA:Maximizing accuracy in global network alignment[J].Bioinformatics,2014,30(20):2931-2940.
[16] Ibragimov R,Malek M,Guo Jiong,et al.GEDEVO:An evolutionary graph edit distance algorithm for biological network alignment[C]//German Conference on Bioinformatics,2013,34:68-79.
[17] Franceschini A,Szklarczyk D,Frankild S,et al.STRING v9.1:Protein-protein interaction networks,with increased coverage and integration[J].Nucleic Acids Research,2013,41(1):808-815.
[18] Von M C,Jensen L J,Snel B,et al.STRING:Known and predicted protein-protein associations,integrated and transferred across organisms[J].Nucleic Acids Research,2005,33:433-437.
[19] Melak T,Gakkhar S.Maximum flow approach to prioritize potential drug targets of Mycobacterium tuberculosis H37Rv from protein-protein interaction network[J].Clinical and Translational Medicine,2015,4(1):61.
[20] Milenkoviæ T,Pržulj N.Uncovering biological network function via graphlet degree signatures[J].Cancer Informatics,2008,6(1):257.
[21] Przulj N.Biological network comparison using graphlet degree distribution[J].Bioinformatics,2007,23(2):177-183.
[22] Milenković T,Ng W L,Hayes W,et al.Optimal network alignment with graphlet degree vectors.[J].Cancer Informatics,2010,9(9):121-137.
[23] Pickl A,Johnsen U,Archer R M,et al.Identification and characterization of 2-keto-3-deoxygluconatekinase and 2-keto-3-deoxygalactonate kinase in the haloarchaeon Haloferax volcanii[J].Fems Microbiology Letters,2014,361(1):76-83.
[24] Ahmed H,Ettema T J G,Tjaden B,et al.The semiphosphorylative Entner-Doudoroff pathway in hyperthermophilic archaea:A re-evaluation[J].Biochemical Journal,2005,390(2):529-540.
[25] Han T K,Zhu Zhiwen,Dao M L.Identification,molecular cloning,and sequence analysis of a deoxyribose aldolase in streptococcus mutans GS-5[J].Current Microbiology,2004,48(3):230-236.
[26] Matthews L R,Vaglio P,Reboul J,et al.Identification of potential interaction networks using sequence-based searches for conserved protein-protein interactions or“Interologs”[J].Genome Research,2001,11(12):2120-2126.
MIAO Mengjun,DING Yanrui.PPI network alignment for study on carbohydrate metabolism of Lactobacillus Plantarum.Computer Engineering andApplications,2018,54(6):49-54.
MIAO Mengjun1,DING Yanrui2
1.School of Internet of Things,Jiangnan University,Wuxi,Jiangsu 214122,China 2.School of Digital Media,Jiangnan University,Wuxi,Jiangsu 214122,China
Protein-Protein Interaction network(PPI network)alignment is an important method for predicting proteinprotein interaction,and analyzing functional differences between different species.In this paper,in order to study the differences of the functional modules of the carbohydrate metabolism in Lactobacillus plantarum WCFS1 and JDM1,the PPI network of Glycolysis module,Pentose phosphate pathway module,Citrate cycle module in WCFS1 and JDM1 are aligned,by Evolutionary Graph Edit Distance Algorithm.Experiment shows that the Edge Correctness of three modules in WCFS1 and JDM1 reaches 93.6%,96%,100%,respectively.It indicates that the topological structure of the carbohydrate metabolism module is very similar.And in pentose phosphate pathway of WCFS1 and JDM1,finding that WCFS1 has protein 2-keto-3-deoxygluconate kinase(kdgK)and JDM1 only has its product 2-keto-3-deoxy-6-phospho-gluconate aldolase(JDM1_0578)no kdgK.In addition,in Glycolysis module,inferring protein pyruvate dehydrogenase complex,E2 component(pdhC)and protein pyruvate kinase(pyk)have interaction.The experimental results show that PPI network alignment can clarify the differences of the carbohydrate metabolism module,topological similarity and can predict the interaction between proteins by aligning the PPI network.
PPI network alignment;Lactobacillus plantarum WCFS1 and JDM1;carbohydrate metabolism functional modules
蛋白质-蛋白质相互作用网络(PPI网络)比对是预测蛋白质相互作用,分析不同物种之间功能差异的重要手段。为研究植物乳杆菌WCFS1和JDM1糖代谢功能模块差异,采用Evolutionary Graph Edit Distance Algorithm算法对两者的糖酵解、戊糖磷酸途径、柠檬酸循环三个模块PPI网络进行比对。实验表明,两者的三个模块边正确性分别达到93.6%、96%、100%,表明其拓扑结构极其相似,戊糖磷酸途径中,WCFS1存在蛋白质2-keto-3-deoxygluconate kinase(kdgK),但JDM1中没有kdgK,却有其产物2-keto-3-deoxy-6-phospho-gluconate aldolase(JDM1_0578)。糖酵解模块中,推测蛋白质pyruvate dehydrogenase complex,E2 component(pdhC)与pyruvate kinase(pyk)具有相互作用。实验表明,PPI网络比对可以阐明两者糖代谢PPI网络的拓扑相似性及模块差异,预测蛋白质之间的相互作用。
PPI网络比对;植物乳杆菌WCFS1和JDM1;糖代谢功能模块
2016-11-15
2017-01-04
1002-8331(2018)06-0049-06
book=54,ebook=59
A
TP399
10.3778/j.issn.1002-8331.1611-0279
国家自然科学基金(No.21541006)。
苗孟君(1991—),女,硕士研究生,研究领域为生物信息,E-mail:15906197927@163.com;丁彦蕊(1976—),女,博士,教授,研究领域为生物信息。
猜你喜欢
江西水产科技(2022年2期)2022-05-17
现代仪器与医疗(2021年1期)2021-06-09
中国循证心血管医学杂志(2020年11期)2020-01-08
中成药(2019年12期)2020-01-04
中华老年口腔医学杂志(2016年4期)2017-01-15
中国医学装备(2016年6期)2016-12-01
软件导刊(2016年9期)2016-11-07
通信电源技术(2016年5期)2016-03-22
陶瓷学报(2015年4期)2015-12-17
应用化工(2015年2期)2015-07-13
