
Dabrafenib, an inhibitor of RIP3 kinase-dependent necroptosis, reduces ischemic brain injury
2018-03-14 07:38ShellyCruzZhaohongQinAlexandreStewartHsiaoHueiChen
中国神经再生研究(英文版) 2018年2期
Shelly A. Cruz, Zhaohong Qin, Alexandre F. R. Stewart, Hsiao-Huei Chen,
1 Ottawa Hospital Research Institute, Ottawa, Canada
2 University of Ottawa, Brain and Mind Institute, Ottawa, Canada
3 University of Ottawa Heart Institute, Ottawa, Canada
4 Canadian Partnership for Storke Recovery, Ottawa, Canada
Introduction
Stroke following ischemic brain injury is a leading cause of death worldwide. Ischemic brain injury resulting from disruption of blood supply to the brain triggers immediate cell death by necrosis, but also activates additional cellular mechanisms,including caspase-dependent apoptosis and caspase-independent necroptosis that propagate a wave of delayed cell death(Tovar-y-Romo et al., 2016).
Release of glutamate from dying neurons and astrocytes triggers excitotoxicity (Schock et al., 2008), a hyper-excitation of glutamatergic receptors that disrupts intracellular Ca2+homeostasis, increases production of reactive oxygen species and release of cytochromce c from mitochondria that activates caspases (Degterev et al., 2003). Injured or dead neurons also activate local brain-resident macrophages (microglia)that secrete chemokines and inflammatory cytokines, including tumor necrosis factor-alpha (TNF-α), to recruit more immune cells in attempt for tissue repairs. However, inflammatory cytokines secreted from immune cells also induce cell death through the activation of the Fas/TNFR family of death-domain receptors (DRs).
Although activation of the caspase family of cysteine proteases is essential to mediate DR signal transduction and execution of apoptosis (Degterev et al., 2003), many studies have shown that caspase inhibitors, including the pancaspase inhibitor zVAD.fmk, fail to block DR-induced cell death (Schulze-Osthoff et al., 1994; Kawahara et al., 1998;Vercammen et al., 1998; Khwaja and Tatton, 1999; Holler et al., 2000; Matsumura et al., 2000; Chan et al., 2003). Emerging evidence points to non-apoptotic caspase-independent cell death pathway called necroptosis, in which DRs activate receptor-interacting protein kinase 1 (RIPK1) and a signaling cascade involving the phosphorylation of RIPK3 by RIPK1(Weinlich et al., 2017). Ischemic brain injury activates necroptotic cell death by activating RIPK3 (Vieira et al., 2014). The RIPK1 inhibitor necrostatin-1 limits experimentally induced ischemic brain injury in mice (Degterev et al., 2005; Degterev et al., 2008), but necrostatin-1 has not yet undergone phase 1 clinical trials for safety and tolerance in humans.
It has emerged that Dabrafenib, an inhibitor of B-raf currently approved for the treatment of melanoma (Hauschild et al., 2012), is also an effective high affinity inhibitor of RIPK3 that blocks TNF-α-induced necroptosis (Li et al., 2014b). In human vascular endothelial cells, Dabrafenib markedly attenuates the activation of TNF-α (Jung et al., 2016). Here, we tested whether Dabrafenib has a neuroprotective effect in an experimentally induced stroke model.
Materials and Methods
Ischemic brain injury by photothrombosis in mice
C57BL6 adult male mice aged 2–3 months, weighing approximately 33 g, were fed with regular chow, and randomly assigned to experimental groups. All procedures were carried out with the approval of the animal care and use committee of the University of Ottawa (OGHRI-49), according to animal use guidelines of the Canadian Council on Animal Care (www.ccac.ca/en/standards/guidelines/). Photothrombosis-induced focal ischemic brain injury was carried out in two-month-old mice, using the protocol of Watson et al. (1985) with the modifications we previously described (Cruz et al., 2017). Mice were anesthesized in an induction chamber ventilated with 5% isoflurane gas (AbbVie Corporation, St-Laurent, Quebec,Canada), transferred to a stereotaxic frame and anesthesia was maintained with 1.5% isoflurane with a nose mask. A midline incision of the scalp exposed a 1 × 1 mm2region of the skull.Rose Bengal (Sigma-Aldrich, Oakville, Ontario, Canada, 10 mg/mL in PBS, made fresh by vortexing and filtered) was administered by i.p. injection (100 mg/kg) and five minutes later, a laser was used to induce thrombosis. A collimated green laser (532 nm wavelength at 20 mW, Beta Instruments, Zaventem, Belgium) was placed 2 cm above the skull, positioned 0.7 mm anteroposterior and 2 mm mediolateral relative to the Bregma to target the sensorimotor cortex and used to illuminate the skull for 10 minutes. The skin wound was closed using cyanoacrylate glue, and mice were monitored until they regained consciousness. Sham operated mice received Rose Bengal and underwent surgery without laser illumination.
One hour after photothrombosis, Dabrafenib (10 mg/kg,Selleck, Houston, TX, USA) or vehicle (dimethyl sulfoxide,DMSO) was delivered by i.p. injection in 150 μL 80% DMSO in saline. Daily maintenance doses of Dabrafenib (5 mg/kg)were administered, prior to sacrifice on day 4.
Quantification of infarction
Mice were sacrificed and perfused with saline followed by 4%paraformaldehyde and brains were isolated and sectioned on a cryostatmicrotome for histology. Infarct volumes were determined by cresyl violet staining to reveal Nissl bodies of living neurons in 10 coronal sections (20 μm thick) sampled every 10 sections over a 2 mm distance overlapping the area of infarction,as previously described (Schock et al., 2008). Infarct volume was calculated by stacking infarct areas in serial sections using the ImageJ software (NIH, Bethesda, MD, USA). Lesion volumes were normalized to brain volumes of the corresponding sections to control for edema. Investigators were blinded to genotype.
Immunofluorescence
Areas surrounding the infarction were visualized with primary antibodies incubated at 4°C overnight to the macrophage marker Iba1 (WAKO Chemicals, Richmond, VA, USA, #019-19741, rabbit anti-rat,1:500 dilution) and neuronal marker NeuN (Millipore Canada Ltd., Etobicoke, Ontario, Canada,MAB377, mouse anti-mouse, 1:500 dilution) by immunofluorescence as previously described (Chen et al., 2007). Cy2 and Cy3 conjugated secondary antibodies were obtained from Jackson Laboratories (Bar Harbour, ME, USA) and used at 1:1,000 dilution. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Iba1-positive cells were imaged on a Zeiss (Oberkoken, Germany) AxioImager Z1 fluorescence microscope and counted in three independent fields at 20× magnification from six sections per mouse.
Bone marrow-derived macrophage cultures
Bone marrow-derived macrophages (BMDM) were cultured for 6 days in HyClone™ Dulbecco’s Modified Eagles Medium(GE Healthcare Life Sciences, Missassauga, Ontario, Canada)conditioned with medium from L926 cells, as we described(Chen et al., 2015) and treated with lipopolysaccharides (LPS;Sigma-Aldrich, Oakville, Ontario, Canada, 100 ng/mL) with or without Dabrafenib (10 μM) for 4 hours. RNA was isolated for quantitative reverse transcription-polymerase chain reaction (qRT-PCR) analysis.
qRT-PCR
Total RNA from BMDM or frozen brain tissue was extracted using the Qiagen (Germantown, MD, USA) RNeasy Mini Kit (Hari et al., 2017). qRT-PCR was conducted as described previously (Pandey et al., 2013; Qin et al., 2015), and the results were normalized to beta-actin. In brief, 1 μg of RNA was used for cDNA synthesis (ABM 5x All-in-one RT MasterMix,#AG490), and 4 μL of 1:4 diluted cDNA was used for qPCR with the following primers: TNF-α: (F) 5′-CCA CCA CGC TCT TCT GTC TAC-3′, (R) 5′-AGG GTC TGG GCC ATA GAA CT-3′ and normalized to beta-actin: (F) 5′-CCT TCT GAC CCA TTC CCA CC, (R) 5′-GCT TCT TTG CAG CTC CTT CG-3′ for cultured BMDM ornormalized to GAPDH: (F)5′-TGT TCC TAC CCC CAA TGT GT-3′, (R) 5′-TGT GAG GGA GAT GCT CAG TG-3′ for brain tissue.
Statistical analysis
All results are presented as the mean ± SEM. For infarct volumes, percentages were normalized by arcsine transformation.For between-group comparisons of fold changes in TNF-α expression, values were normalized by log transformation. Twoway analysis of variance (ANOVA) was used to compare the main effects of treatment (vehicleversusDabrafenib) and time(1 dayversus4 days) and the interaction (treatmentversustime) following photothrombosis on infarct volumes. For experiments in BMDM, two-way ANOVA was used to compare the effects of treatment (vehicleversusDabrafenib) and immune stimulation (with or without LPS challenge) as well as the interaction on TNF-α mRNA levels. Forpost-hocanalysis,the Bonferroni correction was applied for multiple pairwise testing using two-tailed Student’st-test. Differences in means were considered significant atP< 0.05.
Results and Discussion
Dabrafenib (10 mg/kg) administered one hour after photothrombosis-induced focal ischemic injury significantly reduced infarct lesion size in C57Bl6 mice one day after infarction (Figure 1). Two-factor ANOVA revealed a main effect of Dabrafenib (F= 16.458,P= 0.00036) and time (F= 10.131,P= 0.0035) to reduce infarction volume. Administration of a daily maintenance dose of Dabrafenib (5 mg/kg) for 3 days did not further reduce the infarction on day 4. Ischemic injury induced infiltration of Iba1-positive cells (microglia/macrophages), as revealed by immunofluorescence (Figure 2A).Although Dabrafenib treatment did not result in a significant reduction in Iba1+microglia recruitment to the site of injury(Figure 2B), Dabrafenib treatment attenuated up-regulation of TNF-α mRNA levels one day after photothrombosis (Figure 3A, Dabrafenib effect:F= 5.479,P= 0.037; time effect:F=15.412,P= 0.002). This result suggests that the effect of Dabrafenib on reduction of infarct volume is related to attenuated local inflammation after ischemic injury.
To test whether Dabrafenib affects TNF-α activation in macrophages, bone marrow-derived macrophages were stimulated with LPS in the absence or presence of Dabrafenib.Dabrafenib blocked LPS-induced activation of TNF-α expression in macrophages (Figure 3B; Dabrafenib effect:F= 642.54,P= 8.632E-12; LPS effect: 465.39,P= 5.735E-11;interaction:F= 220.49,P= 4.36E-9). Thisin vitrodata may explain why markedly lower TNF-α mRNA level was detected one day after Dabrafenib treatment (10 mg/kg, 1 hour after photothrombosis) despite similar numbers of Iba1-positive microglia/macrophages recruited to the ischemic site. However, we are puzzled by the lack of a sustained effect of Dabrafenib 4 days after stroke. This suggests that the maintenance dose (5 mg/kg/day) we used was not sufficient. It is noteworthy that in humans, Dabrafenib is administered in two 150 mg doses daily (equivalent to 4 mg/kg body weight per day,given a 75 kg average weight) for the treatment of melanoma(Hauschild et al., 2012). On the other hand, in mice, daily oral administration of 10 mg/kg was effective at suppressing tumor growth (King et al., 2013), a dose we also found to be effective when administered intraperitoneally. Future studies are needed to test whether higher maintenance doses of Dabrafenib can further reduce infarct volumes and favor recovery.
Of note, vehicle treated mice showed regression of the infarction volume (expressed as a percentage of brain volume to correct for edema) at 4 days compared to day 1. Regression of infarction volume after photothrombosis has been reported by us (Cruz et al., 2017) and others (Li et al., 2014a). Local inflammation likely contributes to the size of infarction, since we previously showed that mice with sustained inflammation have delayed regression of infarction (Cruz et al., 2017).This would be consistent with the acute anti-inflammatory effect of the higher dose of Dabrafenib we observed 1 day after stroke.
Since the tumour necrosis factor receptor (TNFR) is a major death receptor involved in the activation of necroptosis,we propose a model whereby inhibition of TNF-α production from macrophages/microglia likely accounts for part of the protective effect by Dabrafenib to limit stroke injury (see diagram;Figure 4). Ischemic stroke results in an abrupt deprivation of nutrient supplies that quickly leads to irreversible damage in the core of the affected area. Damage-associated molecular patterns (DAMPS) released from dying cells are sensed by Toll-like receptors (TLRs). TLRs are a receptor family broadly expressed in many cell types including neurons and glia cells. Activation of TLRs triggers gene expression through several transcription factors. Interferon regulatory factors (IRFs) are the main targets of TLRs that initiate the innate immune response. Activation of TLRs/IRFs increases inflammatory cytokine expression, including TNF-α, leading to the recruitment of immune cells, like macrophages, to the site of injury for tissue repair (Kariko et al., 2004). On the other hand, TNF-α activates the TNF receptor on neurons and causes programmed cell death via caspase-dependent and independent pathways (Degterev et al., 2003).
TNF-α further activates IRF1 (Jaruga et al., 2004) that increases expression of RIPK proteins and amplifies TNF-αinduced RIPK3 activation and necroptosis (McComb et al.,2014). Consistent with the activation of TNF-α in ischemic brain injury, IRF1 expression is also elevated in ischemic neurons in mice after experimentally-induced stroke and in post-mortem neurons of stroke patients (Alexander et al.,2003). In line with these observations, mice that lack IRF1 have a smaller infarction after experimental focal brain ischemia (Iadecola et al., 1999).
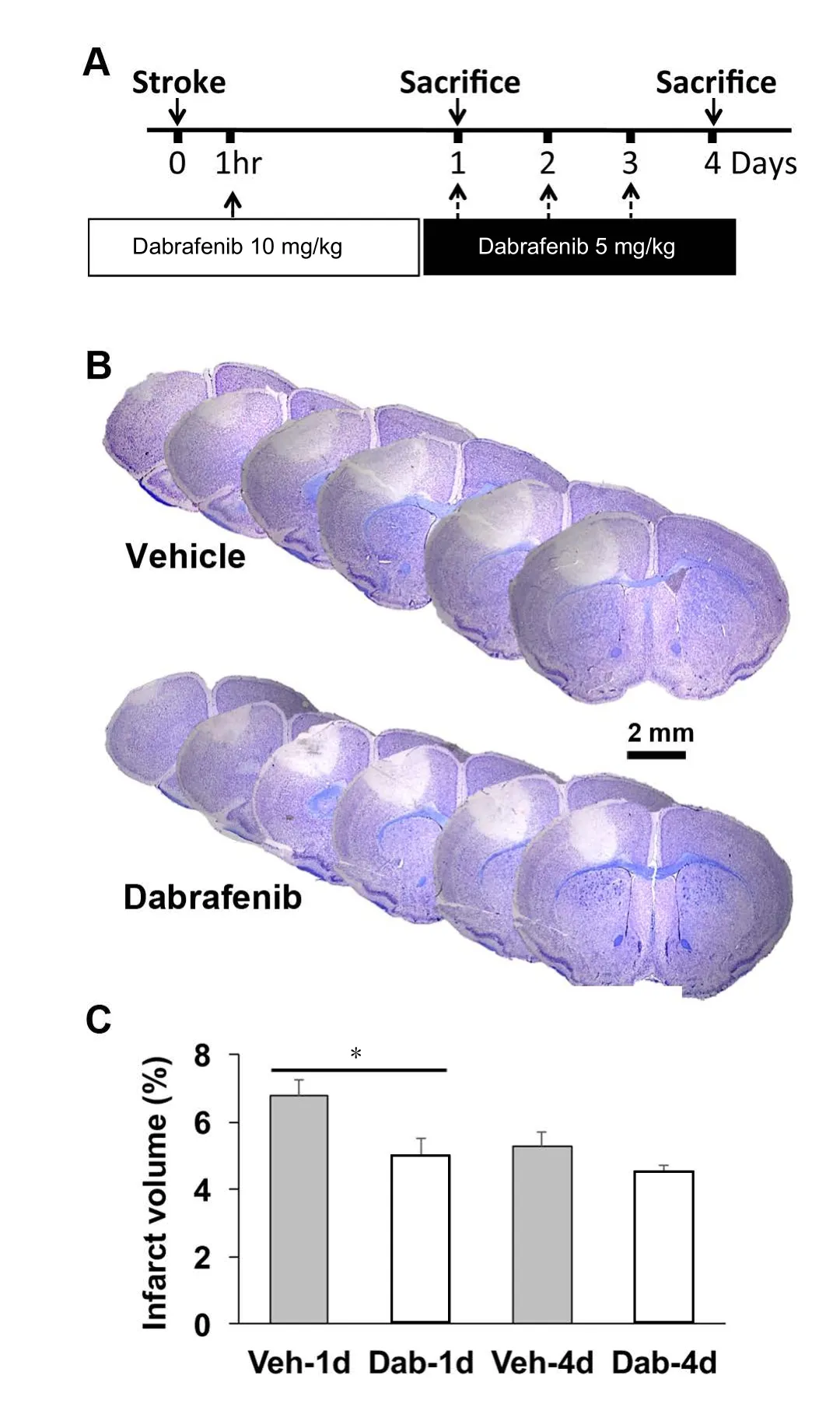
Figure 1 Dabrafenib attenuated ischemic brain injury in mice.
Blocking necroptosis with the RIPK1 inhibitor necrostatin-1was found to be neuroprotective in ischemic brain injury(Degterev et al., 2005). However, necrostatin-1 and related RIPK1 inhibitors also exhibit some toxicity (Takahashi et al.,2012) and most have not undergone clinical trials for safety in humans. Recently, Dabrafenib was identified as a RIPK3 in-hibitor (Li et al., 2014b). Importantly, Dabrafenib has already undergone clinical trials and is an FDA approved drug for the treatment of melanoma in humans (Hauschild et al., 2012).Thus, to the best of our knowledge, our study is the first to demonstrate that Dabrafenib attenuates stroke injury in a preclinical mouse model. Our finding is exciting because it suggests that Dabrafenib could readily be repurposed for stroke therapy.
Whether the effect of Dabrafenib is mediated by its inhibition of RIPK3 remains to be demonstrated. Another RIPK3 inhibitor was recently shown to protect cultured neurons against injury caused by oxygen and glucose deprivation(Fayaz et al., 2016). However, it should be pointed out that ablation of RIPK3 had no effect on brain injury caused by middle cerebral artery occlusion followed by reperfusion (Newton et al., 2016). The discrepancy between the RIPK3 knockout study and our findings could arise from compensatory effects of genetic ablation of RIPK3, or from a difference in stroke models, since photothrombosis does not involve reperfusion.We also cannot exclude that B-raf inhibition by Dabrafenib may itself be beneficial to stroke injury, as has been shown for other B-raf inhibitors (Ahnstedt et al., 2011). Lastly, additional behaviour studies will be required to test whether higher or more frequent doses of Dabrafenib can improve sensorimotor functional recovery after ischemic brain injury.
Author contributions:SAC, ZQ, AFRS and HHC contributed to research design. SAC and ZQ conducted the experiments and acquired data. SAC, AFRS and HHC analyzed the data. AFRS and HHC wrote the manuscript. All authors reviewed and approved the final version of this paper.
Conflicts of interest:None declared.
Financial support:This study was supported by grants from the Heart and Stroke Foundation of Canada (HHC, AFRS), the Canadian Institutes of Health Research (to HHC and AFRS). HHC was also supported by a Mid-Career Investigator Award from the Heart and Stroke Foundation of Ontario.
Research ethics:All procedures were carried out with the approval of the animal care and use committee of the University of Ottawa (OGHRI-49), accord-ing to animal use guidelines of the Canadian Council on Animal Care (www.ccac.ca/en/standards/guidelines/).

Figure 2 Immunofluorescence did not reveal an obvious difference in microglial recruitment between vehicle (Veh) and Dabrafenib(Dab)-treated mice one day after ischemic injury.
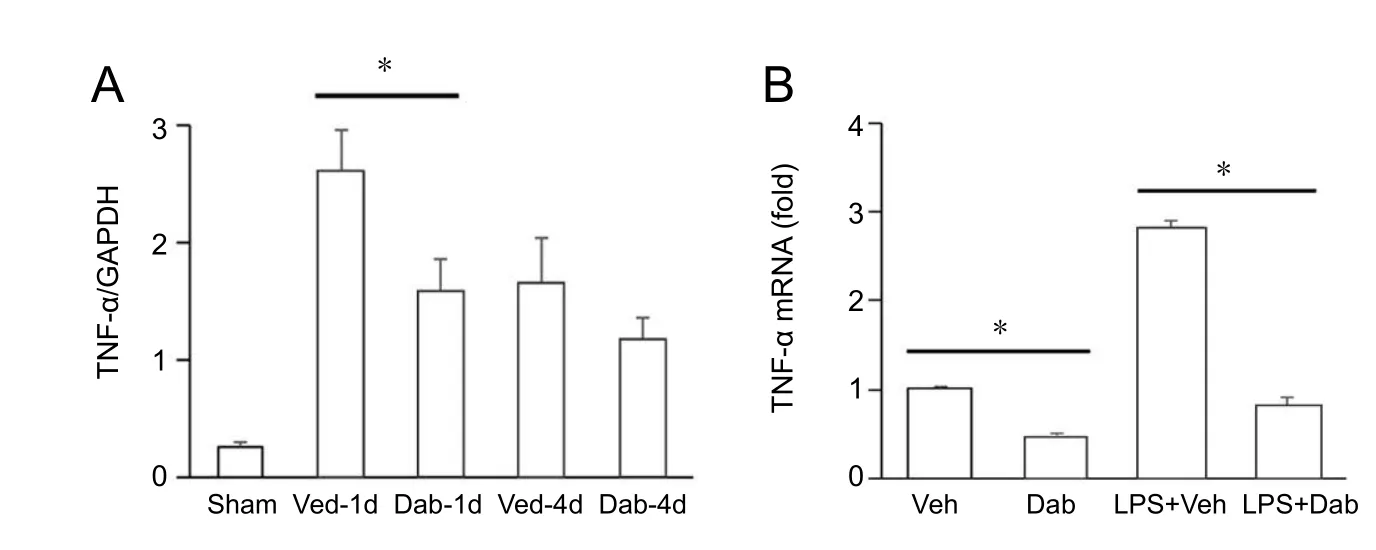
Figure 3 Dabrafenib (Dab) attenuated tumor necrosis factor-alpha (TNF-α) activation in the brain.
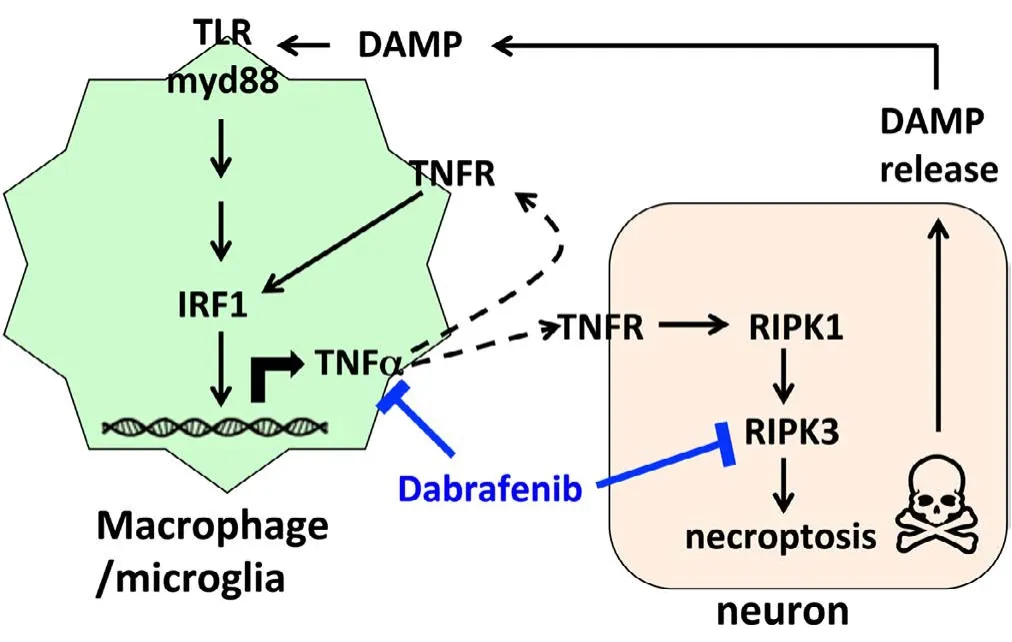
Figure 4 Diagram of Dabrafenib action to block TNF-α-mediated activation of necroptosis induced after ischemic injury.
Data sharing statement:Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under identical terms.
Open peer reviewer:P Aguilera, National Institute of Neurology and Neurosurgery “Manuel Velasco Suárez”, Mexico.
Additional file:Open peer review report 1.
Ahnstedt H, Saveland H, Nilsson O, Edvinsson L (2011) Human cerebrovascular contractile receptors are upregulated via a B-Raf/MEK/ERK-sensitive signaling pathway. BMC Neurosci 12:5.
Alexander M, Forster C, Sugimoto K, Clark HB, Vogel S, Ross ME, Iadecola C (2003) Interferon regulatory factor-1 immunoreactivity in neurons and inflammatory cells following ischemic stroke in rodents and humans. Acta Neuropathol 105:420-424.
Chan FK, Shisler J, Bixby JG, Felices M, Zheng L, Appel M, Orenstein J, Moss B, Lenardo MJ (2003) A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem 278:51613-51621.
Chen HH, Schock SC, Xu J, Safarpour F, Thompson CS, Stewart AF (2007) Extracellular ATP-dependent upregulation of the transcription cofactor LMO4 promotes neuron survival from hypoxia. Exp Cell Res 313:3106-3116.
Chen HH, Keyhanian K, Zhou X, Vilmundarson RO, Almontashiri NA, Cruz S, Pandey NR, Yap NL, Ho T, Stewart CA, Huang H, Hari A, Geoffrion M,McPherson R, Rayner KJ, Stewart AF (2015) IRF2BP2 reduces macrophage inflammation and susceptibility to atherosclerosis. Circ Res 117:671-683.
Cruz SA, Hari A, Qin Z, Couture P, Huang H, Lagace DC, Stewart AFR, Chen HH (2017) Loss of IRF2BP2 in microglia increases inflammation and functional deficits after focal ischemic brain injury. Front Cell Neurosci 11:201.
Degterev A, Boyce M, Yuan J (2003) A decade of caspases. Oncogene 22:8543-8567.
Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD,Mitchison TJ, Moskowitz MA, Yuan J (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1:112-119.
Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D,Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J (2008) Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 4:313-321.
Fayaz SM, Suvanish Kumar VS, Davis CK, Rajanikant GK (2016) Novel RIPK3 inhibitors discovered through a structure-based approach exert post-ischemic neuroprotection. Mol Divers 20:719-728.
Hari A, Cruz SA, Qin Z, Couture P, Vilmundarson RO, Huang H, Stewart AFR, Chen HH (2017) IRF2BP2-deficient microglia block the anxiolytic effect of enhanced postnatal care. Sci Rep 7:9836.
Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M,Rutkowski P, Blank CU, Miller WH Jr, Kaempgen E, Martín-Algarra S,Karaszewska B, Mauch C, Chiarion-Sileni V, Martin AM, Swann S, Haney P, Mirakhur B, Guckert ME, Goodman V, et al. (2012) Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 380:358-365.
Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J (2000) Fas triggers an alternative,caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 1:489-495.
Iadecola C, Salkowski CA, Zhang F, Aber T, Nagayama M, Vogel SN, Ross ME(1999) The transcription factor interferon regulatory factor 1 is expressed after cerebral ischemia and contributes to ischemic brain injury. J Exp Med 189:719-727.
Jaruga B, Hong F, Kim WH, Gao B (2004) IFN-gamma/STAT1 acts as a proinflammatory signal in T cell-mediated hepatitis via induction of multiple chemokines and adhesion molecules: a critical role of IRF-1. Am J Physiol Gastrointest Liver Physiol 287:G1044-1052.
Jung B, Kang H, Lee W, Noh HJ, Kim YS, Han MS, Baek MC, Kim J, Bae JS(2016) Anti-septic effects of dabrafenib on HMGB1-mediated inflammatory responses. BMB Rep 49:214-219.
Kariko K, Weissman D, Welsh FA (2004) Inhibition of toll-like receptor and cytokine signaling--a unifying theme in ischemic tolerance. J Cereb Blood Flow Metab 24:1288-1304.
Kawahara A, Ohsawa Y, Matsumura H, Uchiyama Y, Nagata S (1998)Caspase-independent cell killing by Fas-associated protein with death domain. J Cell Biol 143:1353-1360.
Khwaja A, Tatton L (1999) Resistance to the cytotoxic effects of tumor necrosis factor alpha can be overcome by inhibition of a FADD/caspase-dependent signaling pathway. J Biol Chem 274:36817-36823.
King AJ, Arnone MR, Bleam MR, Moss KG, Yang J, Fedorowicz KE,Smitheman KN, Erhardt JA, Hughes-Earle A, Kane-Carson LS, Sinnamon RH, Qi H, Rheault TR, Uehling DE, Laquerre SG (2013) Dabrafenib; preclinical characterization, increased efficacy when combined with trametinib, while BRAF/MEK tool combination reduced skin lesions. PLoS One 8:e67583.
Li H, Zhang N, Lin HY, Yu Y, Cai QY, Ma L, Ding S (2014a) Histological,cellular and behavioral assessments of stroke outcomes after photothrombosis-induced ischemia in adult mice. BMC Neurosci 15:58.
Li JX, Feng JM, Wang Y, Li XH, Chen XX, Su Y, Shen YY, Chen Y, Xiong B, Yang CH, Ding J, Miao ZH (2014b) The B-Raf(V600E) inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury. Cell Death Dis 5:e1278.
Matsumura H, Shimizu Y, Ohsawa Y, Kawahara A, Uchiyama Y, Nagata S (2000) Necrotic death pathway in Fas receptor signaling. J Cell Biol 151:1247-1256.
McComb S, Cessford E, Alturki NA, Joseph J, Shutinoski B, Startek JB, Gamero AM, Mossman KL, Sad S (2014) Type-I interferon signaling through ISGF3 complex is required for sustained Rip3 activation and necroptosis in macrophages. Proc Natl Acad Sci U S A 111:E3206-3213.
Newton K, Dugger DL, Maltzman A, Greve JM, Hedehus M, Martin-McNulty B, Carano RA, Cao TC, van Bruggen N, Bernstein L, Lee WP, Wu X,DeVoss J, Zhang J, Jeet S, Peng I, McKenzie BS, Roose-Girma M, Caplazi P, Diehl L, et al. (2016) RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ 23:1565-1576.
Pandey NR, Zhou X, Qin Z, Zaman T, Gomez-Smith M, Keyhanian K, Anisman H, Brunel JM, Stewart AF, Chen HH (2013) The LIM domain only 4 protein is a metabolic responsive inhibitor of protein tyrosine phosphatase 1B that controls hypothalamic leptin signaling. J Neurosci 33:12647-12655.
Qin Z, Zhou X, Pandey NR, Vecchiarelli HA, Stewart CA, Zhang X, Lagace DC, Brunel JM, Beique JC, Stewart AF, Hill MN, Chen HH (2015) Chronic stress induces anxiety via an amygdalar intracellular cascade that impairs endocannabinoid signaling. Neuron 85:1319-1331.
Schock SC, Xu J, Duquette PM, Qin Z, Lewandowski AJ, Rai PS, Thompson CS, Seifert EL, Harper ME, Chen HH (2008) Rescue of neurons from ischemic injury by peroxisome proliferator-activated receptor-gamma requires a novel essential cofactor LMO4. J Neurosci 28:12433-12444.
Schulze-Osthoff K, Krammer PH, Droge W (1994) Divergent signalling via APO-1/Fas and the TNF receptor, two homologous molecules involved in physiological cell death. EMBO J 13:4587-4596.
Takahashi N, Duprez L, Grootjans S, Cauwels A, Nerinckx W, DuHadaway JB, Goossens V, Roelandt R, Van Hauwermeiren F, Libert C, Declercq W,Callewaert N, Prendergast GC, Degterev A, Yuan J, Vandenabeele P (2012)Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis 3:e437.
Tovar-y-Romo LB, Penagos-Puig A, Ramirez-Jarquin JO (2016) Endogenous recovery after brain damage: molecular mechanisms that balance neuronal life/death fate. J Neurochem 136:13-27.
Vercammen D, Brouckaert G, Denecker G, Van de Craen M, Declercq W, Fiers W, Vandenabeele P (1998) Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J Exp Med 188:919-930.
Vieira M, Fernandes J, Carreto L, Anuncibay-Soto B, Santos M, Han J, Fernandez-Lopez A, Duarte CB, Carvalho AL, Santos AE (2014) Ischemic insults induce necroptotic cell death in hippocampal neurons through the up-regulation of endogenous RIP3. Neurobiol Dis 68:26-36.
Watson BD, Dietrich WD, Busto R, Wachtel MS, Ginsberg MD (1985) Induction of reproducible brain infarction by photochemically initiated thrombosis. Ann Neurol 17:497-504.
Weinlich R, Oberst A, Beere HM, Green DR (2017) Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol 18:127-136.

- 中国神经再生研究(英文版)的其它文章
- Neuroprotective effects of statins against amyloid βinduced neurotoxicity
- Detection of thinned corticospinal tract and corticoreticular pathway in a patient with a calf circumference discrepancy
- Dyslipidemia modulates Müller glial sensing and transduction of ambient information
- Mitochondrial transplantation strategies as potential therapeutics for central nervous system trauma
- A new direction for Alzheimer’s research
- DNA plasticity and damage in amyotrophic lateral sclerosis