
Neuroprotective effects of statins against amyloid βinduced neurotoxicity
2018-03-14 07:38HsinHuaLiChihLiLinChienNingHuang
中国神经再生研究(英文版) 2018年2期
Hsin-Hua Li, Chih-Li Lin,, Chien-Ning Huang,
1 Institute of Medicine, Chung Shan Medical University, Taichung, Taiwan, China
2 Department of Internal Medicine, Chung Shan Medical University Hospital, Taichung, Taiwan, China
3 Department of Medical Research, Chung Shan Medical University Hospital, Taichung, Taiwan, China
Introduction
Alzheimer’s disease (AD) is a neurodegeneration disorder that is pathologically characterized by cerebral atrophy (particularly within the hippocampus and temporal and parietal lobes), senile plaques, neurofibrillary tangles, and neuronal cell death (Zhang et al., 1989). The clinical features are loss of short-term memory and cognitive function. The pathogenesis of AD is associated with amyloid β (Aβ) peptide and tau hyperphosphorylation, which are a crucial hallmark(Ghiso and Frangione, 2002; Bloom, 2014). Statins have a pathophysiological relationship with AD and brain metabolism abnormalities (Zissimopoulos et al., 2017). Moreover,Aβ accumulation is associated with a number of metabolic brain abnormalities (Sato and Morishita, 2015). Abnormal neuronal metabolism is considered a risk factor for developing AD (Vance, 2012; Sato and Morishita, 2015). As a result,an imbalance in the metabolic status of the brain is believed to be one of the underlying pathophysiologic mechanisms contributing to AD (Kang and Rivest, 2012). In particular,dysregulation of cholesterol homeostasis in the brain considerably increases the risk of developing AD (Vance, 2012).The levels of cholesterol are elevated, which are a central risk factor in the development of AD (Vance, 2012). However, lowering cholesterol levels may benefit the brain of a patient with AD (Vance, 2012). Therefore, cholesterol-lowering medications such as statins are considered to prevent the development of AD. Epidemiological studies indicated that the global use of statins to treat hypercholesterolemia can reduce the risk of AD (Zissimopoulos et al., 2017). The present review highlights the results of brain metabolic abnormalities accelerating the pathogenesis of AD and explains the potential mechanism of statin’s protection against Aβ-induced neurotoxicity.
Cholesterol Homeostasis Abnormalities
Accelerate the Pathogenesis of AD
AD can be classified broadly into two groups: early onset AD and late-onset AD. Early onset AD occurs among individuals < 65 years, whereas late-onset AD affects individuals> 65 years. Although early onset AD only accounts for a small percentage of AD cases, early onset AD is the most severe form, with the majority of cases caused by mutations in one of three genes, namely, the amyloid precursor protein (APP), presenilin 1 (PS1), and presenilin 2 (PS2). By contrast, late-onset AD accounts for the vast majority of AD cases (about 95%). Age is the major risk factor for late-onset AD, and possession of an apolipoprotein ε4 allele (ApoE ε4) is also a risk factor. ApoE ε4 allele-carrying individuals also have an increased risk of developing diabetes, dyslipidemia, hypertension, and hypercholesterolemia. Thus, in the 1990s, high levels of plasma cholesterol and the presence of coronary artery disease were positively correlated with the incidence of AD (Martins et al., 2009).
Imbalanced metabolic status of the brain is believed to be one of the underlying pathophysiologic mechanisms contributing to AD (Kang and Rivest, 2012). To maintain optimal neuronal functions, cholesterol levels are precisely controlled by the brain; altered cholesterol metabolism may contribute to the pathogenesis of neurodegeneration (Nicholson and Ferreira, 2010; Cartocci et al., 2017). Clinical evidence has suggested that dysregulated lipid metabolism may participate in the progression of AD (Vance, 2012; Sato and Morishita, 2015), especially, hypercholesterolemia increases susceptibility to AD (Kang and Rivest, 2012; Vance, 2012;Sato and Morishita, 2015). Increasing evidence suggests that cholesterol plays a role in the pathophysiology of AD.For instance, an elevated serum cholesterol level has been shown to be a risk factor for AD (Wolozin et al., 2000). The presence of ApoE ε4, which is associated with high circulating levels of cholesterol, is a well-established risk factor for developing AD in humans (Jarvik et al., 1994; Gomez-Isla et al., 1996), and cholesterolemia is a frequent finding among patients with AD (Ledesma and Dotti, 2012). As we known the ApoE gene encodes a ~34 kDa protein that serves as a crucial regulator of cholesterol homeostasis throughout the body. Particularly, evidence identifies ApoE is the primary cholesterol carrier in the central nervous system. In humans,ApoE exists as three major alleles including ApoE2, ApoE3,and ApoE4, and inheritance of the ApoE4 allele increases the risk of developing AD. Previously studies have identified that the cholesterol transporter ATP-binding cassette transporter A1 (ABCA1) is a crucial regulator of ApoE levels and lipidation in the brain. Deficiency of ABCA1 leads to a loss of approximately 80% of ApoE in the brain, and the residual 20% that remains is poorly lipidated (Hirsch-Reinshagen et al., 2009). Several independent studies have also shown this poorly lipidated ApoE increases amyloid burden in mouse models of AD, demonstrating that ApoE lipidation by ABCA1 affects key steps in amyloid deposition or clearance. Conversely, robust overexpression of ABCA1 in the brain promotes ApoE lipidation and nearly eliminates the formation of mature amyloid plaques (Hirsch-Reinshagen et al., 2009; Di Paolo and Kim, 2011). These suggest that the lipid binding capacity of ApoE is a major mechanism of its function in the pathogenesis of AD, and imply that increasing ApoE lipidation may display therapeutic importance for this devastating disease (Figure 1). It is known the high cholesterol levels are related to the development of AD neuropathology (Nicholson and Ferreira, 2010). In particular,imbalanced metabolic status of the brain is believed to be one of the most underlying pathophysiologic mechanisms contributing to AD (Kang and Rivest, 2012).
One of the most widely accepted theories of Alzheimer’s pathology is the aggregation of Aβ into extracellular cortical and hippocampal plaques. Aβ denotes peptides of 36–43 amino acids that are crucially involved in AD as the main component of amyloid plaques found in the brains of AD’s patients (Hamley, 2012). The Aβ peptides derive from the APP, which is cleaved by β-secretase and γ-secretase to yield Aβ. Aβ molecules can aggregate to form flexible soluble oligomers which may exist in several forms such as monomeric, oligomers and fibrillary forms. Although the normal functional of Aβ is not well understood (Hiltunen et al., 2009), several potential studies have been indicated that Aβ-caused neurotoxicity, including oxidative stress (Li et al., 2016), regulation of cholesterol transport (Igbavboa et al., 2009) and anti-microbial activity, which potentially associated with Aβ-induced inflammatory activity. Therefore, transgenic AD mice studies found that mitochondrial cholesterol overloading exacerbates Aβ-induced inflammation and neurotoxicity in AD (Fernández et al., 2009). A recent study indicated that the involvement of cholesterol in APP metabolism is suggested by the fact that cholesterol is a membrane lipid and Aβ is produced by intra-membrane cleavage of APP. Therefore, cholesterol may increase the activity of β-secretase or γ-secretase enzymes that generate Aβ from APP, decrease the flux of APP through the non-amyloidogenic α-secretase pathway, and affect various non-amyloid factors such as local inflammation or tau metabolism(Cole et al., 2005; Shinohara et al., 2014). As a result, the potential mechanisms for cholesterol’s apparent adverse effect on the development of AD act on APP primarily at the cell surface. However, the accumulation of Aβ protein in the brain is a slow process that takes several years before manifesting its neurotoxicity (Spires-Jones and Hyman, 2014).The presence of amyloid plaques in elderly subjects without cognitive impairment suggests that the accumulation of the peptide by itself is not the only causative condition of neuronal damage; for unknown reasons, Aβ becomes progressively toxic in the brain of patients with AD (Geula et al., 1998; Fjell and Walhovd, 2012). The brain is the organ with the highest cholesterol content, the majority of which stems fromde novosynthesis (Pfrieger and Ungerer, 2011).Notably, high cholesterol levels have recently been found to be significantly elevated in patients with either vascular dementia or AD, and a positive correlation has been reported(Nina et al., 2011). Some study reported that isolated mitochondria from brain or cortical neurons of transgenic mice overexpressing sterol regulatory element binding protein 2 (SREBP-2) or Niemann-Pick type C1 (NPC1) knock-out mice, which contribute to polygenic hypercholesterolaemia,exhibited mitochondrial cholesterol accumulation, mitochondrial glutathione (mGSH) depletion and increased susceptibility to Abeta1–42-induced oxidative stress and release of apoptogenic proteins. Similar findings were observed in pharmacologically GSH-restricted rat brain mitochondria,while selective mGSH depletion sensitized human neuronal and glial cell lines to Aβ1–42-mediated cell death (Fernández et al., 2009).In vitrostudies have demonstrated that secretion of cholesterol leads to neuronal damage (Zhang and Liu, 2015), but the potential molecular mechanisms for cholesterol’s apparent adverse effect on Aβ-induced neurotoxicity are unclear.
Statin Prevents Cholesterol-Accelerated AD Pathogenesis
Several lines of evidence suggest that dysregulated lipid metabolism may participate in the progression of AD (Sato and Morishita, 2015). In particular, imbalances in the cholesterol homeostasis of the brain considerably increase the risk of developing AD (Vance, 2012). Brain cholesterol levels increase the susceptibility of neurons to Aβ toxicity,thereby revealing a possible role in triggering AD (Nicholson and Ferreira, 2010). Therefore, elevated cholesterol levels are a major risk factor for AD. And, lowered brain cholesterol levels may benefit patients with AD. Given these concepts, cholesterol-lowering medications, such as statins,are considered to help prevent or lower the risk of AD. Epidemiological studies highly suggest that statins can globally reduce the risk of AD (Jick et al., 2000; Wolozin et al., 2000;Zissimopoulos et al., 2017). The global use of statins to treat hypercholesterolemia has led to the hope that statins may prove useful in treating or preventing AD.Statins,also known as 3-hydroxy-3-methylglutaryl coenzyme A(HMG-CoA) reductase inhibitors, are a class of lipid-lowering medications (Stancu and Sima, 2001). Statins are principally used in the treatment of hypercholesterolemia(Catapano et al., 2016). They include medications such as atorvastatin, fluvastatin, lovastatin, pitavastatin, pravastatin,mevastatin, rosuvastatin, cerivastatin, and simvastatin. The synthetic statins atorvastatin, rosuvastatin, cerivastatin, and fluvastatin appear to be as efficacious as the natural ones,such as mevastatin lovastatin, pravastatin, and simvastatin(Chong et al., 2001). Statins have been categorized into two types: lipophilic and hydrophilic (Fong, 2014). Lipophilic statins, which include simvastatin, lovastatin, atorvastatin,and cerivastatin, can easily cross the blood brain barrier.By contrast, hydrophilic statins, which include fluvastatin,mevastatin, and pravastatin, are a group of compounds that block cholesterol biosynthesisviacompetitive inhibition of HMG-CoA reductase. HMG-CoA reductase is the rate-limiting enzyme that catalyzes the conversion of HMG-CoA to mevalonate in cholesterol biosynthesis (Chong et al., 2001;Fong, 2014). Excess free cholesterol in the cell is converted into cholesteryl esters by the enzyme sterol O-acyltransferase 1 (ACAT1; also known as acyl CoA:cholesterol acyltransferase 1), followed by accumulation in intracellular lipid droplets or efflux through the plasma membrane into the extra-cellular environment (Chang et al., 2006). Increasing levels of cholesteryl esters enhances Aβ release in cultured cells, whereas pharmacological inhibition of ACAT1 leads to the reduction of both Aβ and cholesteryl ester. Genetic ablation of ACAT1 reduces both Aβ pathology and cognitive impairments in a mouse model of AD (Bryleva et al.,2010). Together, these suggested that the balance between free cholesterol and cholesterol esters is a key parameter controlling amyloidogenesis (Figure 1). The bioavailability of statins varies from less than 5% with simvastatin to approximately 60% with cerivastatin. They inhibit the enzyme HMG-CoA reductase, which plays a central role in the production of cholesterol and other sterols (Chong et al., 2001).This mechanism of action and its effect on lipids (low-density lipoprotein (LDL) cholesterol reduction, triglyceride reduction, and high-density lipoprotein cholesterol elevation)are well established (Vaughan et al., 2000; Stancu and Sima,2001). Statins lower cholesterol production and promote hepatic removal of serum LDL cholesterol. In addition to their conventional use in lowering cholesterol in humans,statins have been widely used for the treatment of a variety of conditions.
Several lines of evidence have indicated that statins can reduce AD pathology in both clinical studies and animal studies (Sano et al., 2011; Geifman et al., 2017; Zissimopoulos et al., 2017). Clinical studies found that cholesterol may be implicated in the pathogenesis of AD, and statins administered in midlife might prevent AD in late life (Geifman et al., 2017; Zissimopoulos et al., 2017). However, Sano et al.(2011) reported that statins do not benefit the progression of symptoms in individuals with mild to moderate AD despite a significant reduction in cholesterol. A recent re-analysis of AD patient-level data from failed clinical trials suggested that the use of statins may benefit all patients with AD with potentially greater therapeutic efficacy than those homozygous for ApoE4 (Geifman et al., 2017). However, increasing evidence suggests that the reduction in AD risk varied across statin molecules, sex, and race/ethnicity (Zissimopoulos et al., 2017), and there is a lower prevalence of diagnosed probable AD in patients taking 2 different statin such as lovastatin and pravastatin (Wolozin et al., 2000) (Table 1).In summary, statins may prevent the pathology of AD. Significant emerging evidence has linked cholesterol, Aβ, and AD, and several studies have shown a reduced risk for AD and dementia in populations treated with statins. In experimental settings in animal models, statins were found to reduce the Aβ level and tau hyperphosphorylation in the brain(Ostrowski et al., 2007; Kurinami et al., 2008; Shinohara et al., 2010; Kurata et al., 2011; Papadopoulos et al., 2014). Aβ reduction is associated with a reduction in the APP-carboxyl terminal fragment (APP-CTF) by statin treatment (Shinohara et al., 2010). Statins reduce brain Aβ levels by increasing APP-CTF trafficking through isoprenylation inhibition (Shinohara et al., 2010). Moreover, statin enhances Aβ clearance by upregulating LDL receptor-related protein 1 expression in brain microvessels (Shinohara et al., 2010). Hence, prevention of Aβ-induced memory impairment by fluvastatin has been associated with reduced brain Aβ accumulation and oxidative stress in an AD mouse model (Kurinami et al., 2008). Some studies indicated that atorvastatin and pitavastatin can improve cognitive function and reduce senile plaque and phosphorylated tau in aged APP mice (Kurata et al., 2011). Handattu et al. (2009) also showed that pravastatin significantly ameliorates Aβ burden in the hippocampus and improves cognitive function in an AD mouse model. As a result, statin can significantly attenuate cholesterol-accelerated AD pathogenesis in animal and AD patient studies(Table 1). Mechanistically, whether the regimen of statins used in this trial influences pathogenic mechanisms of AD in the brain or affected biomarkers of amyloid, tau, or other neuropathologies remains unclear.
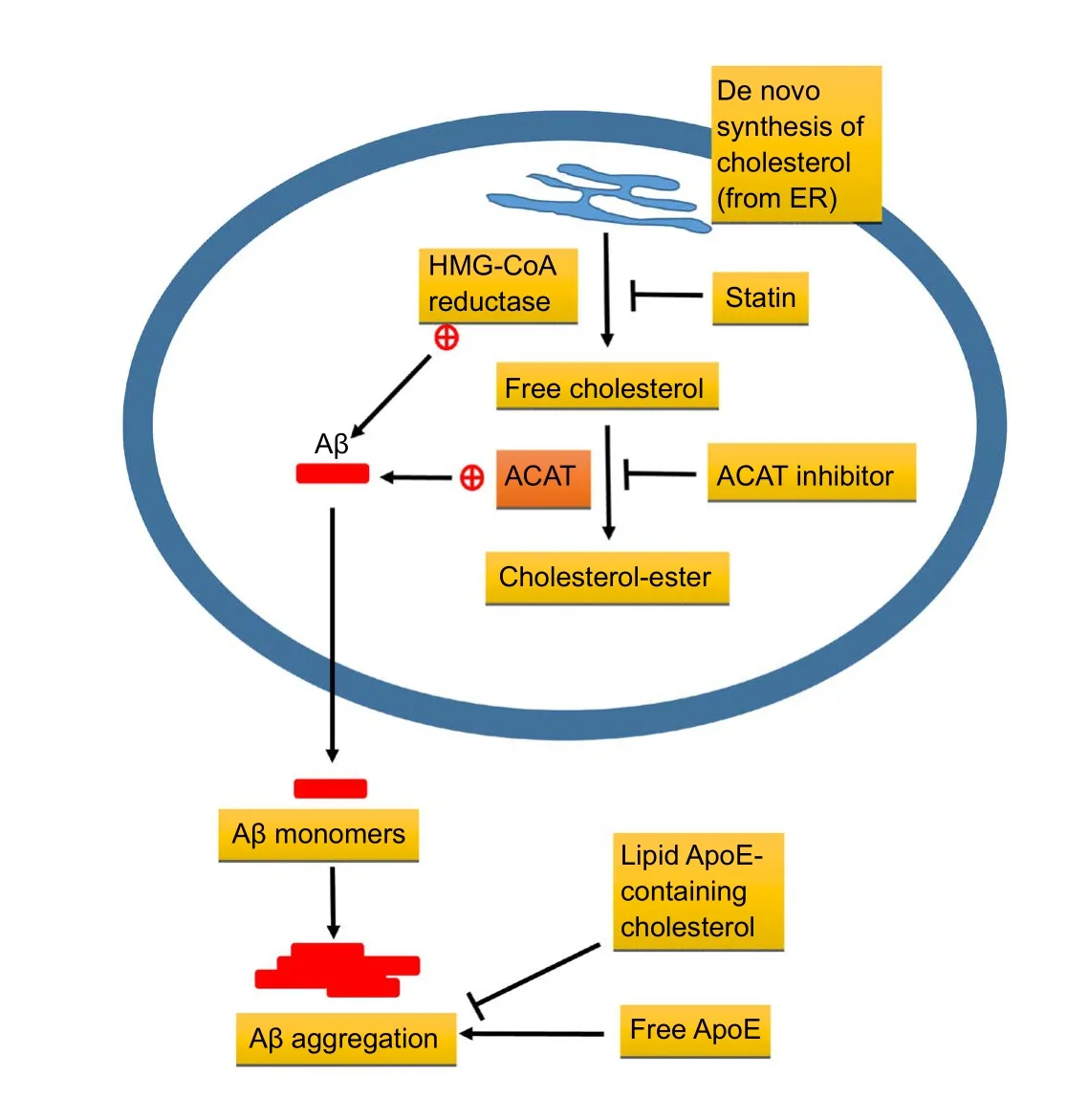
Figure 1 Contribution of cholesterol to biogenesis and degradation of amyloid β (Aβ).
Statin Prevents Cholesterol-Promoted Aβ Deposit Formation and Neurotoxicity
Clinical studies demonstrated that elevated levels of seru m or cerebrospinal fluid cholesterol are linked to the production of Aβ peptides and the development of AD (Papassotiropoulos et al., 2003; Guasti et al., 2008). Aβ is a 38 to 43 amino acid peptide that is derived from the APP through sequential cleavages by β- and γ-secretase enzyme activities. The cleavage site for another APP processing enzyme,α-secretase lies within the Aβ sequence and thus precludes Aβ formation. Excess Aβ is believed to be a main contributor to the dysfunction and degeneration of neurons that occurs in AD (Thinakaran and Koo, 2008). A previous study reported that statin may reduce the cellular cholesterol level of living hippocampal neurons by 70%, and the formation of Aβ is completely inhibited while the generation of a nonamyloidogenic secreted form is unperturbed. This indicates that depletion of cholesterol in the brain inhibits the generation of Aβ in hippocampal neurons (Simons et al., 1998). Moreover,some study indicated that treatment with lovastatin results in a higher expression of the α-secretase ADAM 10, and inhibit the generation of Aβ in neural cell lines (Kojro et al., 2001).Zandl-Lang et al. (2018) also demonstrated the use of simvastatin displays intriguing effects among cellular cholesterol metabolism, Aβ production metabolism, and clearance at the blood–brain interface. Statins also promote the degradation of extracellular Aβ peptide by microgliaviastimulating exosome-associated insulin-degrading enzyme secretion (Tamboli et al., 2010) (Table 2). Thus, statins indeed attenuate cholesterol-accelerated Aβ production (Figure 1).
Several lines of evidence suggest that dysregulated lipid metabolism may also participate in the pathogenesis of AD.Epidemiologic studies reveal that elevated mid-life plasma cholesterol levels may be associated with an increased risk of AD, and the statin use may significantly reduce the prevalence of AD. Cellular studies have shown that intracellular cholesterol markedly affects the processing of APP into Aβ peptides. A large body of data suggest that statins prevent Aβ-induced cell death by decreasing cholesterol levels (Longenberger and Shah, 2011; Geifman et al., 2017;Kornelius et al., 2017). In fact, Aβ can induce the production of pro-inflammatory cytokines interleukin-1β (IL-1β),tumor necrosis factor-α (TNF-α), and interleukin-6 (IL-6)from activated microglia (Chong et al., 2001) leading to the overload of inflammatory cytokines in the hippocampus(Geifman et al., 2017). For example, the administration of atorvastatin ameliorates cognitive deficits, depresses inflammatory responses, improves long-term potentiation impairment, and prevents Aβ-induced neurotoxicity in cultured hippocampal neurons. These protective functions of atorvastatin involve the pathway of reducing farnesyl pyrophosphate (Zhao et al., 2016). Atorvastatin also attenuates the production of IL-1β, IL-6, and TNF-α in the hippocampus of an Aβ1-42-induced rat model of AD (Corrao et al., 2013). Moreover, simvastatin has been demonstrated to cause a shift in cytokine production from proinflammatory to anti-inflammatory response (Barbosa et al., 2017).Simvastatin also protects Aβ-induced neuron cell death in hippocampal dentate gyrus, which may improve spatial cognitive function in animal studies and clinical studies(Longenberger and Shah, 2011) (Table 3). Thus, HMG-CoA reductase inhibitors (statins) have various pleiotropic effects such as reducing Aβ-induced neuroinflammation, and Aβ-induced neuronal cell death. Although the Aβ-induced neurotoxicity remain controversial, dysregulation of calcium homeostasis and oxidative stress are likely to be a major mechanism that mediates Aβ toxicity. In mixed cultures containing hippocampal neurons and astrocytes, neurotoxic Aβ cause sporadic cytosolic calcium signals in astrocytes but not in neurons, initiating a cascade that ends in neuronal death (Catapano et al., 2016). Considering that mevastatin is a common group of drugs clinically prescribed to treat high cholesterol, we proposed that it may also exert neuroprotective effects by decreasing intracellular cholesterol levels (Kornelius et al., 2017). Actually, high exogenous cho-
lesterol levels may render neurons vulnerable to Aβ-induced neuronal cell death (Mendoza-Oliva et al., 2013). Several studies have indicated that statins can attenuate Aβ-induced neuronal cell apoptosisin vivoandin vitro(Papassotiropoulos et al., 2003; Guasti et al., 2008; Fernández et al., 2009;Handattu et al., 2009). The extracellular accumulation of Aβ is one of the characteristic neuropathological hallmarks of AD in neuritic plaques. Experimental data indicate that different molecular forms of Aβ affect a wide array of neuronal and glial functions, and thereby lead to neuronal death in the nervous system. Martins et. al. demonstrated that atorvastatin prevents the spatial learning and memory deficits by upregulating antioxidant systems mainly in an AD mice model (Martins et al., 2015). Similarly, our recent study reported that mevastatin prevents Aβ-induced cell death by decreasing cholesterol levels (Kornelius et al., 2017) (Table 3). This indicates statin may play a beneficial role in Aβ-induced neurotoxicity through reducing cholesterol levels.
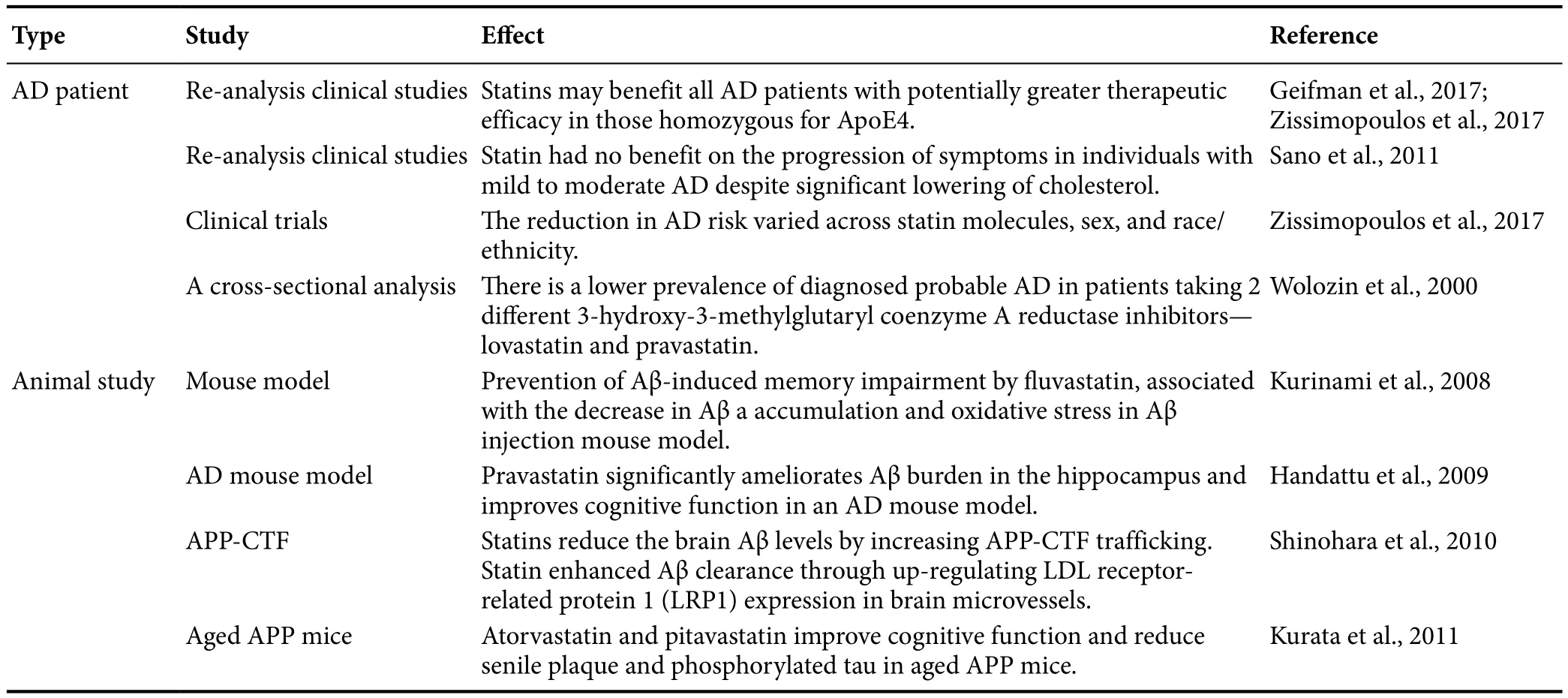
Table 1 Statins reduce AD pathology in clinical studies and animal studies
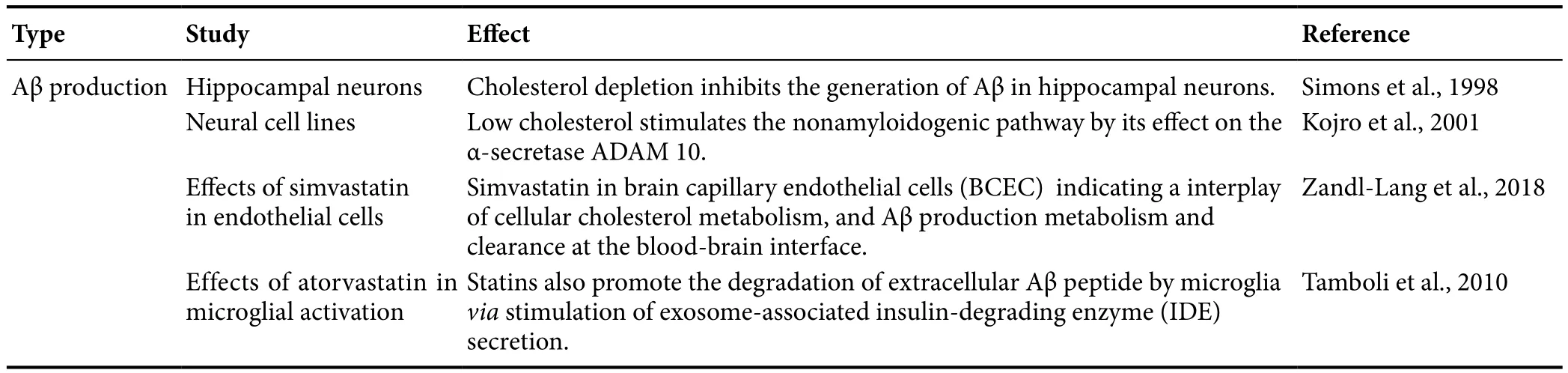
Table 2 Statins attenuate cholesterol-accelerated amyloid β (Aβ) production.

Table 3 Statins attenuate cholesterol-promoted Aβ-induced neurotoxicity
Statins Attenutates Aβ-Induced Neuronal Apoptosis Through Activation of Akt and/or AMP-Activated Protein Kinase (AMPK)Signaling
The dysregulation of lipid metabolism may be initiated or accelerated in the progression of AD pathology (Sato and Morishita, 2015). In particular, dysregulation of cholesterol homeostasis in the brain substantially increases the risk of developing AD (Vance, 2012). This finding indicates that dysregulated lipid homeostasis in the brain can be induced by an increase in Aβ levels or production of longer species of Aβ, such as Aβ42 and Aβ43 peptides (Vance, 2012; Sato and Morishita, 2015). The raised Aβ levels increase neurotoxicity including oxidative stress, mitochondria dysfunction and neuronal cell apoptosis. Previously research demonstrated that Aβ-induced cell death can be enhanced by inhibition of neuronal insulin signaling (Li et al., 2016). In fact, there is widening recognition that AD is closely linked to a state of relative insulin resistance in the brain, so-called “type III diabetes” (de la Monte et al., 2006). It is known that AD is a neurodegenerative disorder defined at the molecular level by the presence of neurofibrillary tangles (NFTs) and insoluble Aβ plaques. NFTs are composed of hyper-phosphorylated forms of the microtubule-associated protein tau, whereas Aβ is derived from the proteolytic cleavage of APP (Hardy, 2006). Tau proteins are essential in assembly as well as maintenance of the structural integrity of microtubules (Hooper et al., 2008). However, Tau is abnormally hyper-phosphorylated and aggregated in AD. Aβ can stimulate hyper-phosphorylation of the Tau protein, which is the main event responsible for NFT formation in AD brains(Hooper et al., 2008). Therefore, neuronal insulin signaling are often dysregulated in AD brain (Messier and Teutenberg, 2005). In normal brain, insulin promotes glucose utilization, energy metabolism, and neuronal survival through PI3K/Akt signaling. Akt is an important regulator of cell survival and apoptosis (Freude et al., 2009). Thus, Akt is activated by PI3K and then phosphorylates glycogen synthase kinase-3β (GSK-3β) activation. Downregulation of PI3K/Akt signaling causes GSK-3β activity increase and leads to Tau hyper-phosphorylation, the main component of NFT(Hooper et al., 2008; Freude et al., 2009). It has been shown that Akt activation may play a therapeutic role in neurodegenerative diseases such as AD (Kitagishi et al., 2014).Therefore, dysregulated lipid homeostasis can be induced Aβ production, which may accelerate Aβ toxicity and leaded to neuronal apoptosis.
Epidemiological studies suggested that statin use is associated with a decreased incidence of AD. Particularly,some studies have reported that statin protects against Aβ-induced neurotoxicity and apoptosis through Akt activation. For example, atorvastatin and pitavastatin reduced the level of oxidative stress, as revealed by the presence of 4-hydroxy-2-nonenal (4-HNE) and advanced glycation end products (AGEs) in AD mouse brains by improving insulin/Akt signaling in AD mouse brains (Kurata et al.,2013). Simvastatin also prevents Aβ-impaired neurogenesis in hippocampal dentate gyrus through cascading PI3K-Akt and increasing BDNFviareduction in farnesyl pyrophosphate (Wang et al., 2015). Moreover, lovastatin exhibits neuroprotective benefits in preventing neurodegeneration by activating the Akt pathway and inhibits downstream GSK-3β activity (Lin et al., 2016). Our results also indicated that mevastatin attenuates Aβ-induced cell death by upregulating insulin signaling, which can be mediated through the repression of GSK-3β and tau phosphorylation (Kornelius et al., 2017). These findings suggested that GSK3β may trigger phosphorylated tau aggregation and lead to cell apoptosis,whereas statin effectively represses Aβ-induced tau pathology and apoptosis by retuning impaired neuronal insulin signaling (Figure 2).
Discussion
Previously studies demonstrated that statins activate AMPK signaling (Sun et al., 2006). AMPK signaling plays an important role in the coordination of cellular energy and metabolic status in various organs (Lin et al., 2017). AMPK is a trimeric enzyme comprising a catalytic α-subunit and regulatory β, γ-subunits (Stapleton et al., 1997). It was first identified as an upstream kinase that phosphorylates and hence inactivates HMG-CoA reductase and acetyl-CoA carboxylase (ACC), the key enzymes controlling cholesterol/isoprenoid and fatty acid biosynthesis, respectively. AMPK can function as a fuel gauge to regulate the homeostasis of energy in the form of glucose and fatty acids in skeletal muscles, liver, and adipocytes (Kahn et al., 2005). Recent findings suggest that the fuel-sensing mechanism of AMPK is also present in the hypothalamus in regulation of food intake and energy expenditure (Minokoshi et al., 2004). The involvement of AMPK in diabetes mellitus is demonstrated by insulin resistance, with associated high levels of plasma glucose and low levels of insulin in mice with ablated AMPK(Viollet et al., 2003). Previous study reported that statins activate endothelial nitric oxide (NO) synthase (eNOS) with increased NO production, which has been suggested to be mediated through the phosphoinositide 3-kinase (PI3K)/Akt pathway (Sun et al., 2006). Some studies indicated that a sustained decrease in AMPK activity accompanies insulin resistance, whereas AMPK activation increases insulin sensitivity (Steinberg and Kemp, 2009; Salminen et al., 2011). In addition, AMPK activity can reduce Aβ-induced neurotoxicity and restore neuronal insulin signaling(Kornelius et al., 2015). The activated AMPK is one of the possible mechanisms underlying the proposed neuroprotection of statins against Aβ-induced neurotoxicity, rather than directly to their cholesterol-lowering effects. Given that statins can effectively increase activation of AMPK, they may also improve brain insulin action and exert protective effects against AD-impaired insulin signaling (Kornelius et al., 2017). Considering the important roles of Aβ-induced cell death and insulin resistance in AD pathogenesis, these observations unveil a potential neuroprotective mechanism by statin through restoration of AMPK and insulin signaling(Figure 2).
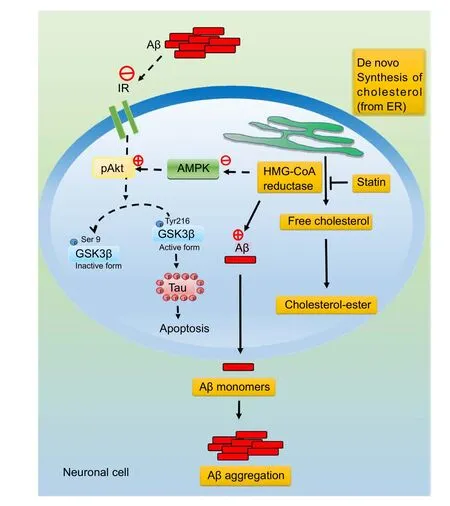
Figure 2 Statins attenuates amyloid β (Aβ)-induced neuronal apoptosis through activation of Akt and/or AMP-activated protein kinase (AMPK) signaling.
Conclusion
AD is the most common form of neurodegenerative dementia. The overproduction or reduced clearance of Aβ peptides in the brain are thought to be central in the pathogenesis of AD. Understanding the variations in the body’s metabolism that can influence brain Aβ levels is important for the development of therapies to reduce the incidence of AD.Although some licensed drugs are used to improve certain symptoms, no drug treatment can provide a cure for AD.AD may be a brain-specific form of metabolism abnormality disease, but the underlying mechanism remains largely unknown. However, dysregulation of cholesterol homeostasis in the brain notably increases the risk of developing AD. In particular, epidemiological studies indicated that the global use of statins to treat hypercholesterolemia can reduce the risk of AD. The above findings indicate that dysregulation of brain cholesterol metabolism by impaired brain insulin action may provide a novel mechanism of altered brain and neuronal function, which represents a potential mechanistic association between insulin resistance and cholesterol lowering treatment by statins. However, the precise mechanism of action for statin-mediated neuroprotection remains to be fully elucidated. Taken together, this review provides important insights of statins and lipid homeostasis on Aβ-induced neurotoxicity. Accordingly, attenuation neurodegeneration by targeting statin-mediated neuronal insulin signaling may lead to novel diagnostic or therapeutic strategies against AD in future.
Author contributions:All authors contributed equally to literature review, text conception, manuscript writing, reviewer contributions/comments, and manuscript finalization.
Conflicts of interest:None declared.
Financial support:This work was supported by the grants from the Ministry of Science and Technology of Taiwan, China (MOST 105-2314-B-013-MY3 and MOST 106-2320-B-040-021-MY3).
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under identical terms.
Open peer review reports:
Reviewer 1:Jérôme Braudeau, AgenT, France.
Comments to authors:The author pertinently summarize the potential interest of the statins use in order to counteract or slow down AD. Despite the fact that this interest is not new, the potential beneficial effect of statins is yet controversy. The review smartly highlighted the potential for statin use resulting from both in vivo and in vitro studies.
Reviewer 2:Sandra I. Mota, University of Coimbra, Portugal.
Comments to authors:The proposed review aims to summarize the potential role of statins, normally used as anti-cholesterol treatment, in AD and their impact on amyloid-beta neurotoxicity. The theme is interesting since there is no recent review about it. However, the article as is,should be improved in a consequent way.
Barbosa CP, Bracht L, Ames FQ, de Souza Silva-Comar FM, Tronco RP, Bersani-Amado CA (2017) Effects of ezetimibe, simvastatin, and their combination on inflammatory parameters in a rat model of adjuvant-induced arthritis. Inflammation 40:717-724.
Bloom GS (2014) Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol 71:505-508.
Bryleva EY, Rogers MA, Chang CC, Buen F, Harris BT, Rousselet E,Seidah NG, Oddo S, LaFerla FM, Spencer TA, Hickey WF, Chang TY (2010) ACAT1 gene ablation increases 24(S)-hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD. Proc Natl Acad Sci U S A 107:3081-3086.
Cartocci V, Servadio M, Trezza V, Pallottini V (2017) Can cholesterol metabolism modulation affect brain function and behavior? J Cell Physiol 232:281-286.
Catapano AL, Graham I, De Backer G, Wiklund O, Chapman MJ,Drexel H, Hoes AW, Jennings CS, Landmesser U, Pedersen TR,Reiner Z, Riccardi G, Taskinen MR, Tokgozoglu L, Verschuren WM, Vlachopoulos C, Wood DA, Zamorano JL (2016) 2016 ESC/EAS Guidelines for the Management of Dyslipidaemias: The Task Force for the Management of Dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS)Developed with the special contribution of the European Assocciation for Cardiovascular Prevention & Rehabilitation (EACPR).Atherosclerosis 253:281-344.
Chang TY, Chang CC, Ohgami N, Yamauchi Y (2006) Cholesterol sensing, trafficking, and esterification. Annu Rev Cell Dev Biol 22:129-157.
Chong PH, Seeger JD, Franklin C (2001) Clinically relevant differences between the statins: implications for therapeutic selection. Am J Med 111:390-400.
Cole SL, Grudzien A, Manhart IO, Kelly BL, Oakley H, Vassar R (2005)Statins cause intracellular accumulation of amyloid precursor protein, beta-secretase-cleaved fragments, and amyloid beta-peptide via an isoprenoid-dependent mechanism. J Biol Chem 280:18755-18770.
Corrao G, Ibrahim B, Nicotra F, Zambon A, Merlino L, Pasini TS,Catapano AL, Mancia G (2013) Long-term use of statins reduces the risk of hospitalization for dementia. Atherosclerosis 230:171-176.
de la Monte SM, Tong M, Lester-Coll N, Plater M Jr, Wands JR (2006)Therapeutic rescue of neurodegeneration in experimental type 3 diabetes: relevance to Alzheimer’s disease. J Alzheimers Dis 10:89-109.
Di Paolo G, Kim TW (2011) Linking lipids to Alzheimer’s disease:cholesterol and beyond. Nat Rev Neurosci 12:284-296.
Fernández A, Llacuna L, Fernandez-Checa JC, Colell A (2009) Mitochondrial cholesterol loading exacerbates amyloid beta peptide-induced inflammation and neurotoxicity. J Neurosci 29:6394-6405.
Fjell AM, Walhovd KB (2012) Neuroimaging results impose new views on Alzheimer’s disease--the role of amyloid revised. Mol Neurobiol 45:153-172.
Fong CW (2014) Statins in therapy: understanding their hydrophilicity, lipophilicity, binding to 3-hydroxy-3-methylglutaryl-CoA reductase, ability to cross the blood brain barrier and metabolic stability based on electrostatic molecular orbital studies. Eur J Med Chem 85:661-674.
Freude S, Schilbach K, Schubert M (2009) The role of IGF-1 receptor and insulin receptor signaling for the pathogenesis of Alzheimer’s disease: from model organisms to human disease. Curr Alzheimer Res 6:213-223.
Geifman N, Brinton RD, Kennedy RE, Schneider LS, Butte AJ (2017)Evidence for benefit of statins to modify cognitive decline and risk in Alzheimer’s disease. Alzheimers Res Ther 9:10.
Geula C, Wu CK, Saroff D, Lorenzo A, Yuan M, Yankner BA (1998)Aging renders the brain vulnerable to amyloid beta-protein neurotoxicity. Nat Med 4:827-831.
Ghiso J, Frangione B (2002) Amyloidosis and Alzheimer’s disease.Adv Drug Deliv Rev 54:1539-1551.
Gomez-Isla T, West HL, Rebeck GW, Harr SD, Growdon JH, Locascio JJ, Perls TT, Lipsitz LA, Hyman BT (1996) Clinical and pathological correlates of apolipoprotein E epsilon 4 in Alzheimer’s disease. Ann Neurol 39:62-70.
Guasti L, Marino F, Cosentino M, Maio RC, Rasini E, Ferrari M, Castiglioni L, Klersy C, Gaudio G, Grandi AM, Lecchini S, Venco A(2008) Prolonged statin-associated reduction in neutrophil reactive oxygen species and angiotensin II type 1 receptor expression: 1-year follow-up. Eur Heart J 29:1118-1126.
Hamley IW (2012) The amyloid beta peptide: a chemist’s perspective.Role in Alzheimer’s and fibrillization. Chem Rev 112:5147-5192.
Handattu SP, Garber DW, Monroe CE, van Groen T, Kadish I, Nayyar G, Cao D, Palgunachari MN, Li L, Anantharamaiah GM (2009)Oral apolipoprotein A-I mimetic peptide improves cognitive function and reduces amyloid burden in a mouse model of Alzheimer’s disease. Neurobiol Dis 34:525-534.
Hardy J (2006) A hundred years of Alzheimer’s disease research. Neuron 52:3-13.
Hiltunen M, van Groen T, Jolkkonen J (2009) Functional roles of amyloid-beta protein precursor and amyloid-beta peptides: evidence from experimental studies. J Alzheimers Dis 18:401-412.
Hirsch-Reinshagen V, Burgess BL, Wellington CL (2009) Why lipids are important for Alzheimer disease? Mol Cell Biochem 326:121-129.
Hooper C, Killick R, Lovestone S (2008) The GSK3 hypothesis of Alzheimer’s disease. J Neurochem 104:1433-1439.
Igbavboa U, Sun GY, Weisman GA, He Y, Wood WG (2009) Amyloid beta-protein stimulates trafficking of cholesterol and caveolin-1 from the plasma membrane to the Golgi complex in mouse primary astrocytes. Neuroscience 162:328-338.
Jarvik GP, Austin MA, Fabsitz RR, Auwerx J, Reed T, Christian JC,Deeb S (1994) Genetic influences on age-related change in total cholesterol, low density lipoprotein-cholesterol, and triglyceride levels: longitudinal apolipoprotein E genotype effects. Genet Epidemiol 11:375-384.
Jick H, Zornberg GL, Jick SS, Seshadri S, Drachman DA (2000) Statins and the risk of dementia. Lancet 356:1627-1631.
Kahn BB, Alquier T, Carling D, Hardie DG (2005) AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Meta 1:15-25.
Kang J, Rivest S (2012) Lipid metabolism and neuroinflammation in Alzheimer’s disease: a role for liver X receptors. Endocr Rev 33:715-746.
Kitagishi Y, Nakanishi A, Ogura Y, Matsuda S (2014) Dietary regulation of PI3K/AKT/GSK-3beta pathway in Alzheimer’s disease.Alzheimers Res Ther 6:35.
Kojro E, Gimpl G, Lammich S, Marz W, Fahrenholz F (2001) Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha-secretase ADAM 10. Proc Natl Acad Sci U S A 98:5815-5820.
Kornelius E, Li HH, Peng CH, Hsiao HW, Yang YS, Huang CN, Lin CL (2017) Mevastatin promotes neuronal survival against Aβ-induced neurotoxicity through AMPK activation. Metab Brain Dis 32:1999-2007.
Kornelius E, Lin CL, Chang HH, Li HH, Huang WN, Yang YS, Lu YL,Peng CH, Huang CN (2015) DPP-4 inhibitor linagliptin attenuates Aβ-induced cytotoxicity through activation of AMPK in neuronal cells. CNS Neurosci Ther 21:549-557.
Kurata T, Miyazaki K, Morimoto N, Kawai H, Ohta Y, Ikeda Y, Abe K(2013) Atorvastatin and pitavastatin reduce oxidative stress and improve IR/LDL-R signals in Alzheimer’s disease. Neurol Res 35:193-205.
Kurata T, Miyazaki K, Kozuki M, Panin VL, Morimoto N, Ohta Y,Nagai M, Ikeda Y, Matsuura T, Abe K (2011) Atorvastatin and pitavastatin improve cognitive function and reduce senile plaque and phosphorylated tau in aged APP mice. Brain Res 1371:161-170.
Kurinami H, Sato N, Shinohara M, Takeuchi D, Takeda S, Shimamura M, Ogihara T, Morishita R (2008) Prevention of amyloid beta-induced memory impairment by fluvastatin, associated with the decrease in amyloid beta accumulation and oxidative stress in amyloid beta injection mouse model. Int J Mol Med 21:531-537.
Ledesma MD, Dotti CG (2012) Peripheral cholesterol, metabolic disorders and Alzheimer’s disease. Front Biosci (Elite Ed) 4:181-194.
Li HH, Lin SL, Huang CN, Lu FJ, Chiu PY, Huang WN, Lai TJ, Lin CL (2016) miR-302 attenuates amyloid-beta-induced neurotoxicity through activation of Akt signaling. J Alzheimers Dis 50:1083-1098.
Lin CH, Lin HI, Chen ML, Lai TT, Cao LP, Farrer MJ, Wu RM,Chien CT (2016) Lovastatin protects neurite degeneration in LRRK2-G2019S parkinsonism through activating the Akt/Nrf pathway and inhibiting GSK3beta activity. Hum Mol Genet 25:1965-1978.
Lin Z, Zhang Z, Jiang X, Kou X, Bao Y, Liu H, Sun F, Ling S, Qin N,Jiang L, Yang Y (2017) Mevastatin blockade of autolysosome maturation stimulates LBH589-induced cell death in triple-negative breast cancer cells. Oncotarget 8:17833-17848.
Longenberger J, Shah ZA (2011) Simvastatin and other HMG-CoA reductase inhibitors on brain cholesterol levels in Alzheimer’s disease.Curr Alzheimer Res 8:434-442.
Martins WC, dos Santos VV, dos Santos AA, Vandresen-Filho S, Dal-Cim TA, de Oliveira KA, Mendes-de-Aguiar CB, Farina M, Prediger RD, Viola GG, Tasca CI (2015) Atorvsastatin prevents cognitive deficits induced by intracerebroventricular amyloid-beta1-40 administration in mice: involvement of glutamatergic and antioxidant sytems. Neurotox Res 28:32-42.
Mendoza-Oliva A, Ferrera P, Arias C (2013) Interplay between cholesterol and homocysteine in the exacerbation of amyloid-beta toxicity in human neuroblastoma cells. CNS Neurol Disord Drug Targets 12:842-848.
Messier C, Teutenberg K (2005) The role of insulin, insulin growth factor, and insulin-degrading enzyme in brain aging and Alzheimer’s disease. Neural Plast 12:311-328.
Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, Mu J, Foufelle F, Ferre P, Birnbaum MJ, Stuck BJ, Kahn BB (2004)AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 428:569-574.
Nicholson AM, Ferreira A (2010) Cholesterol and neuronal susceptibility to beta-amyloid toxicity. Cogn Sci (Hauppauge) 5:35-56.
Nina E, Shepardson, Ganesh M, Shankar, Dennis J, Selkoe (2011)Cholesterol and statins in Alzheimer’s disease: II. Review of human trials and recommendations. Arch Neurol 68:1385-1392.
Ostrowski SM, Wilkinson BL, Golde TE, Landreth G (2007) Statins reduce amyloid-beta production through inhibition of protein isoprenylation. J Biol Chem 282:26832-26844.
Papadopoulos P, Tong XK, Hamel E (2014) Selective benefits of simvastatin in bitransgenic APPSwe,Ind/TGF-beta1 mice. Neurobiol Aging 35:203-212.
Papassotiropoulos A, Streffer JR, Tsolaki M, Schmid S, Thal D, Nicosia F, Iakovidou V, Maddalena A, Lutjohann D, Ghebremedhin E,Hegi T, Pasch T, Traxler M, Bruhl A, Benussi L, Binetti G, Braak H, Nitsch RM, Hock C (2003) Increased brain beta-amyloid load,phosphorylated tau, and risk of Alzheimer disease associated with an intronic CYP46 polymorphism. Arch Neurol 60:29-35.
Pfrieger FW, Ungerer N (2011) Cholesterol metabolism in neurons and astrocytes. Prog Lipid Res 50:357-371.
Salminen A, Hyttinen JM, Kaarniranta K (2011) AMP-activated protein kinase inhibits NF-κB signaling and inflammation: impact on healthspan and lifespan. J Mol Med (Berl) 89:667-676.
Sano M, Bell KL, Galasko D, Galvin JE, Thomas RG, van Dyck CH,Aisen PS (2011) A randomized, double-blind, placebo-controlled trial of simvastatin to treat Alzheimer disease. Neurology 77:556-563.
Sato N, Morishita R (2015) The roles of lipid and glucose metabolism in modulation of beta-amyloid, tau, and neurodegeneration in the pathogenesis of Alzheimer disease. Front Aging Neurosci 7:199.
Shinohara M, Sato N, Shimamura M, Kurinami H, Hamasaki T, Chatterjee A, Rakugi H, Morishita R (2014) Possible modification of Alzheimer’s disease by statins in midlife: interactions with genetic and non-genetic risk factors. Front Aging Neurosci 6:71.
Shinohara M, Sato N, Kurinami H, Takeuchi D, Takeda S, Shimamura M, Yamashita T, Uchiyama Y, Rakugi H, Morishita R (2010) Reduction of brain beta-amyloid (Abeta) by fluvastatin, a hydroxymethylglutaryl-CoA reductase inhibitor, through increase in degradation of amyloid precursor protein C-terminal fragments (APP-CTFs)and Abeta clearance. J Biol Chem 285:22091-22102.
Simons M, Keller P, De Strooper B, Beyreuther K, Dotti CG, Simons K(1998) Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc Natl Acad Sci U S A 95:6460-6464.
Spires-Jones TL, Hyman BT (2014) The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 82:756-771.
Stancu C, Sima A (2001) Statins: mechanism of action and effects. J Cell Mol Med. 5:378-387.
Stapleton D, Woollatt E, Mitchelhill KI, Nicholl JK, Fernandez CS,Michell BJ, Witters LA, Power DA, Sutherland GR, Kemp BE (1997)AMP-activated protein kinase isoenzyme family: subunit structure and chromosomal location. FEBS Lett 409:452-456.
Steinberg GR, Kemp BE (2009) AMPK in health and disease. Physiol Rev 89:1025-1078.
Sun W, Lee TS, Zhu M, Gu C, Wang Y, Zhu Y, Shyy JY (2006) Statins activate AMP-activated protein kinase in vitro and in vivo. Circulation 114:2655-2662.
Tamboli IY, Barth E, Christian L, Siepmann M, Kumar S, Singh S,Tolksdorf K, Heneka MT, Lutjohann D, Wunderlich P, Walter J(2010) Statins promote the degradation of extracellular amyloid{beta}-peptide by microglia via stimulation of exosome-associated insulin-degrading enzyme (IDE) secretion. J Biol Chem 285:37405-37414.
Thinakaran G, Koo EH (2008) Amyloid precursor protein trafficking,processing, and function. J Biol Chem 283:29615-29619.
Vance JE (2012) Dysregulation of cholesterol balance in the brain: contribution to neurodegenerative diseases. Dis Model Mech 5:746-755.
Vaughan CJ, Gotto AM Jr, Basson CT (2000) The evolving role of statins in the management of atherosclerosis. J Am Coll Cardiol 35:1-10.
Viollet B, Andreelli F, Jorgensen SB, Perrin C, Flamez D, Mu J, Wojtaszewski JF, Schuit FC, Birnbaum M, Richter E, Burcelin R, Vaulont S (2003) Physiological role of AMP-activated protein kinase(AMPK): insights from knockout mouse models. Biochem Soc Trans 31:216-219.
Wang C, Chen T, Li G, Zhou L, Sha S, Chen L (2015) Simvastatin prevents β-amyloid(25-35)-impaired neurogenesis in hippocampal dentate gyrus through α7nAChR-dependent cascading PI3K-Akt and increasing BDNF via reduction of farnesyl pyrophosphate.Neuropharmacology 97:122-132.
Wolozin B, Kellman W, Ruosseau P, Celesia GG, Siegel G (2000)Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol 57:1439-1443.
Zandl-Lang M, Fanaee-Danesh E, Sun Y, Albrecher NM, Gali CC,Cancar I, Kober A, Tam-Amersdorfer C, Stracke A, Storck SM,Saeed A, Stefulj J, Pietrzik CU, Wilson MR, Bjorkhem I, Panzenboeck U (2018) Regulatory effects of simvastatin and apoJ on APP processing and amyloid-beta clearance in blood-brain barrier endothelial cells. Biochim Biophys Acta 1863:40-60.
Zhang H, Sternberger NH, Rubinstein LJ, Herman MM, Binder LI,Sternberger LA (1989) Abnormal processing of multiple proteins in Alzheimer disease. Proc Natl Acad Sci U S A 86:8045-8049.
Zhang J, Liu Q (2015) Cholesterol metabolism and homeostasis in the brain. Protein Cell 6:254-264.
Zhao L, Chen T, Wang C, Li G, Zhi W, Yin J, Wan Q, Chen L (2016)Atorvastatin in improvement of cognitive impairments caused by amyloid β in mice: involvement of inflammatory reaction. BMC Neurol 16:18.
Zissimopoulos JM, Barthold D, Brinton RD, Joyce G (2017) Sex and race differences in the association between statin use and the incidence of Alzheimer disease. JAMA Neurol 74:225-232.

- 中国神经再生研究(英文版)的其它文章
- Dyslipidemia modulates Müller glial sensing and transduction of ambient information
- Detection of thinned corticospinal tract and corticoreticular pathway in a patient with a calf circumference discrepancy
- Contributions of neurotropic human herpesviruses herpes simplex virus 1 and human herpesvirus 6 to neurodegenerative disease pathology
- Mitochondrial transplantation strategies as potential therapeutics for central nervous system trauma
- A new direction for Alzheimer’s research
- DNA plasticity and damage in amyotrophic lateral sclerosis