
一测多评法同时测定柴胡药渣总皂苷中3种次生柴胡皂苷含量
2018-03-04 09:30
中国民族民间医药 2018年24期
河南科技大学化工与制药学院,河南 洛阳 471003
柴胡是一味常用中药,具有和解表里,升阳举陷的作用[1]。汤剂是中医临床用药的主要形式,以水为溶剂的煎煮法是含柴胡成药生产工艺中较为常用的一种的提取方法。本课题组在对涉及柴胡煎煮的相关工艺进行研究时,如柴胡口服液的制备,发现柴胡煎煮过后的药渣中仍然含有大量的皂苷,具有较高的再利用价值。基于此,建立了柴胡药渣中皂苷的提取富集工艺,并从中制备了柴胡皂苷的提取物。该提取物中所含的皂苷不同与柴胡药材中所含的原生柴胡皂苷,如柴胡皂苷a、c、d,而是以次生柴胡皂苷为主,如柴胡皂苷h、b2和b1(分别为柴胡皂苷c、d、a的转化产物)[2-4]。以上述3种次生皂苷为指标建立柴胡药渣皂苷提取物的质量标准时,发现上述柴胡皂苷标准品价格较高,或不易得到,造成所制定的标准在实际应用中存在较大的不便。“一测多评”法,即测定1个成分的含量,利用该成分与其他成分间的校正因子,来实现对多个成分含量的监控,为多指标质量控制标准的实际应用带来便利[5]。
基于此,本文以柴胡皂苷b2为对照品,建立其与柴胡皂苷b1、h的相对校正因子,并用校正因子计算柴胡皂苷的含量。同时以t检验法对相对校正因子的计算值和外标法实测值进行比较,并对不同品牌色谱柱、高效液相色谱仪以及不同实验室的试验结果进行考察,综合评价一测多评法在以柴胡药渣总皂苷质量控制中应用的准确性和科学性。
1 仪器与试药
1.1 仪器 Shimazu LC-15C高效液相色谱仪;LC-Solution 色谱工作站(日本岛津公司);Waters Alliance2695-2487高效液相色谱仪(Empower 2 色谱工作站,美国Waters公司);RE-52AA旋转蒸发仪(郑州长城科工贸有限公司);KQ500DE型数控超声波清洗器(昆山市超声仪器有限公司);FA2004B型电子分析天平(上海佑科仪器仪表有限公司);Hypersil-C18色谱柱(250mm×4.6mm,5μm),WondaSil C18 色谱柱(4.6 mm×250 mm,5 μm),Waters Xbridge C18色谱柱(4.6 mm×250 mm,5 μm)。
1.2 材料 柴胡药渣总皂苷提取物(自制,4批);柴胡皂苷b2、b1、h(自制,经MS、1H-NMR 和13C-NMR确定结构,HPLC 色谱峰面积归一化法测得纯度均HPLC面积归一化法测定其纯度高于98%),柴胡皂苷h(批号:16111514,宝鸡市辰光生物科技有限公司);甲醇(色谱纯,DIKMA公司),哇哈哈纯净水,其它试剂均为分析纯。
2 方法与结果
2.1 一测多评法方法学考察
2.1.1 色谱条件 Hypersil-C18色谱柱(250mm×4.6mm,5μm);流动相:乙腈(A)-水(B)为流动相,梯度洗脱(0~70min,22%→50%);流速:1.0mL·min-1;柱温:35℃;检测波长:254nm;进样量:10.0μL。理论塔板数按照柴胡皂苷b2峰计算应不低于6000。色谱图见图1。
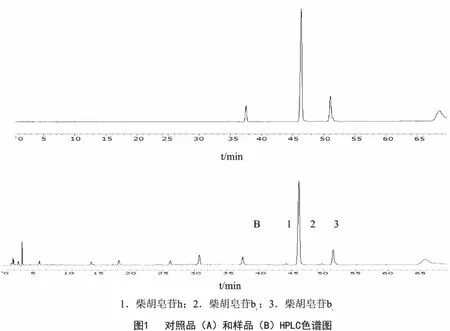
2.1.2 对照品溶液的制备 精密称取柴胡皂苷h、b1、b2对照品适量置于50 mL的量瓶中,配置成浓度分别为0.03、0.1、0.24 mg·mL-1的混合对照品溶液Ⅰ,分别精密吸取上述混合对照品溶液1.5、1.0、0.5、0.2 mL ,置于4个不同的2 mL量瓶中,用甲醇定容,摇匀,作为混合对照品溶液Ⅱ~Ⅴ,于4℃冰箱中保存,备用。
2.1.3 供试品溶液的制备 称取柴胡药渣总皂苷提取物5 mg,精密称定,置于10 mL的容量瓶中,加甲醇溶解,并定容至刻度,摇匀,用0.45 μm微孔滤膜过滤,即得。
2.1.4 线性范围 取“2.1.2”项下混合对照品溶液Ⅰ~Ⅴ,分别精密吸取10 μL注入高效液相色谱仪。按照“2.2.1”项下色谱方法分析,并记录峰面积。以进样量X为横坐标与峰面积Y(A×10-5)为纵坐标,进行线性回归,得柴胡皂苷b1、b2、h的回归方程及线性范围,即柴胡皂苷b1Y=11.93X+0.1188 (r=0.9993),线性范围0.10~1.0 μg;柴胡皂苷b2Y=14.33X+3.233(r=0.9997),线性范围0.24~2.40 μg;柴胡皂苷hY=9.2438X-0.1945(r=0.9996),线性范围0.03~0.30 μg。
2.1.5 精密度试验 精密吸取Ⅳ号混合对照品溶液10 μL,连续进样6次,记录柴胡皂苷b2、b1、h的峰面积,其峰面积的RSD分别为2.10%、1.50%、1.01%。表明仪器精密度良好。
2.1.6 稳定性实验 取同一供试品溶液10 μL分别于1、2、4、6、8、10 h进样测定, 记录柴胡皂苷b2、b1、h峰面积,其峰面积的RSD分别为2.32%, 1.89%和2.30%。表明供试品溶液在10 h内稳定不变。
2.1.7 重复性试验 称取同一批柴胡药渣皂苷富集物5 mg,共5份,精密称定,按供试品溶液处理方法制备样品并测定, 柴胡皂苷h、b1、b2含量的RSD(n=5)分别为,2.82%、2.32%、2.95%,表明重复性良好。
2.1.9 加样回收率 称取已知含量的柴胡皂苷富集物适量5份,每份2.5mg,精密称定,分别加入适量的柴胡皂苷b2、b1、h对照品,按照供试品溶液的处理方法制备样品并依法测定,计算加样回收率,柴胡皂苷b2、b1、h的平均加样回收率为99.7%、99.7%、RSD(n=5)分别为1.03%、1.17%、2.10%。表明实验方法准确度符合要求。
2.2 相对校正因子的计算 在“2.1.1”项下确定的HPLC 色谱分析条件下,精密吸取“2.2.2”项下5 个浓度的混合对照品溶液,分别进样10 μL。以柴胡皂苷b2为内标,根据相对响应因子计算公式fr=(An×Ws)/(As×Wn) ,其中,fr,A,W 分别为相对校正因子、峰面积及成分质量,下标n,s 分别代表待测成分和内参成分[6-8],分别计算柴胡皂苷b1、h的相对响应因子。结果见表1。
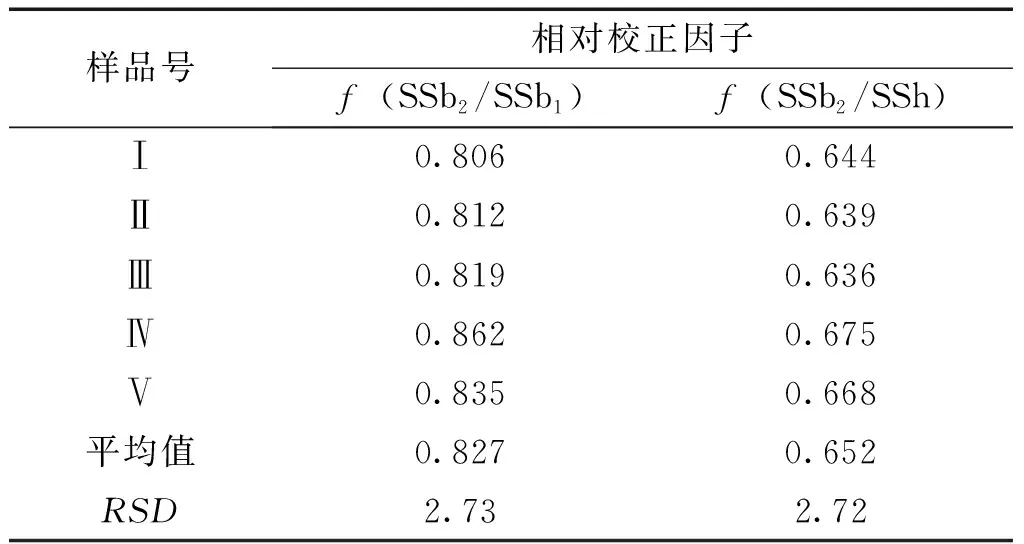
表1 次生柴胡皂苷b1和h的相对校正因子
注:SS-柴胡皂苷
2.3 一评多测法耐用性和系统适应性评价
2.3.1 不同进样量对形影因子的影响 在Shimazu LC-15C、WondaSil C18、流速1.0 mL·min-1下,取“2.1.2”项下对照品溶液Ⅳ,分别进样2、5、10、15、20 μL,计算fssb2/ssb1、fssb2/ssh,考察不同进样量对相对响应因子的影响,结果各成分相对响应因子重现性良好,RSD 值分别为1.11%、3.20%。
2.3.2 色谱柱和高效液相色谱仪考察 实验考察了Shimazu LC-15C和Waters Alliance2695-2487两种高效液相色谱系统和Hypersil-C18(250mm×4.6mm,5μm),WondaSil C18(4.6 mm×250 mm,5 μm),Waters Xbridge C18(4.6 mm×250 mm,5 μm)3种色谱柱。结果见表2。
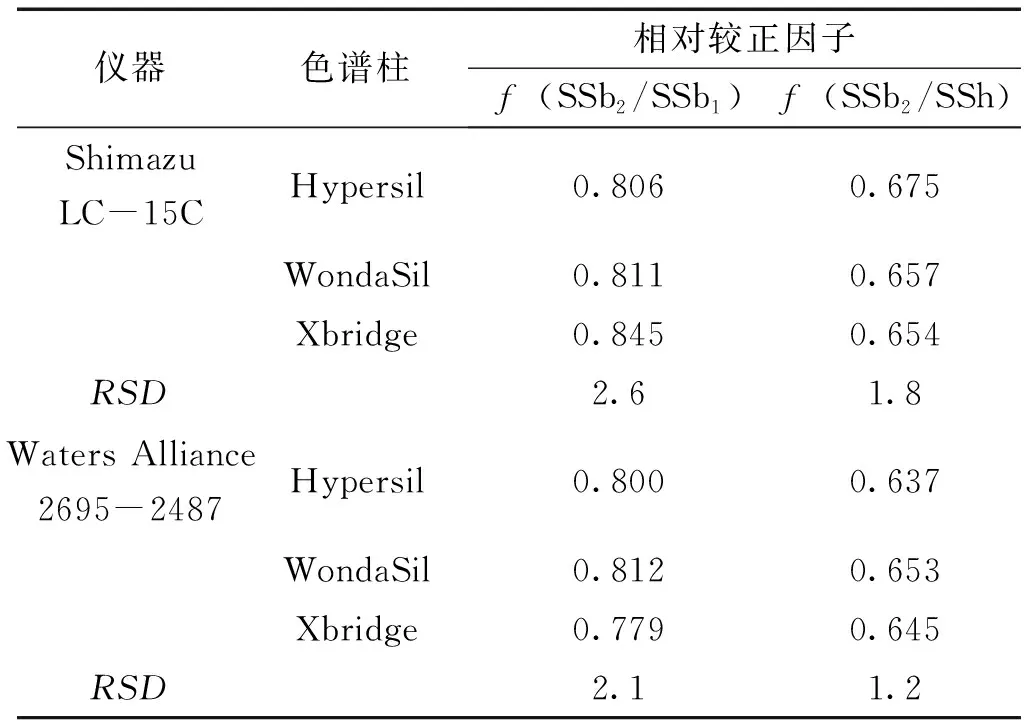
表2 不同仪器和不同色谱柱测得的校正因子
2.3.3 待测组分色谱峰的定位 采用相对保留时间值作为定位标准,计算不同色谱仪和色谱柱的相对保留时间。结果RSD<5%,表明不同色谱仪和色谱柱下相对保留时间波动较小,重现性较好。结果见表3。
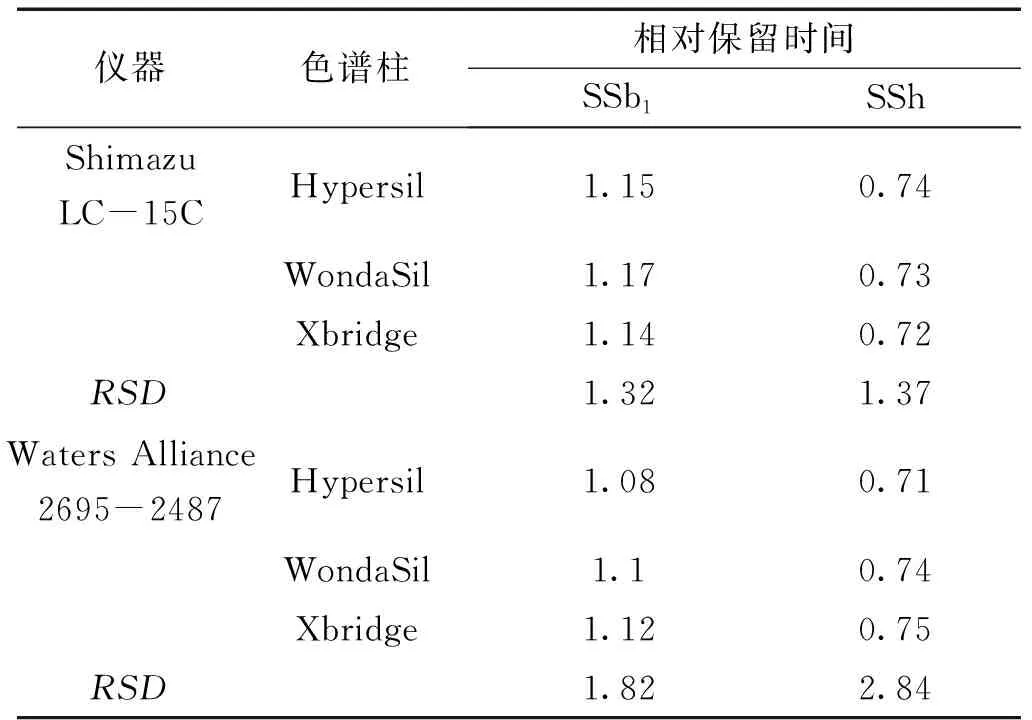
表3 不同色谱仪和色谱柱测得的相对保留时间
2.3.5 实验室考察 取“2.1.2”项下对照品溶液Ⅳ,对建立的一评多测分析方法在两个实验室进行复核实验。结果见表4。
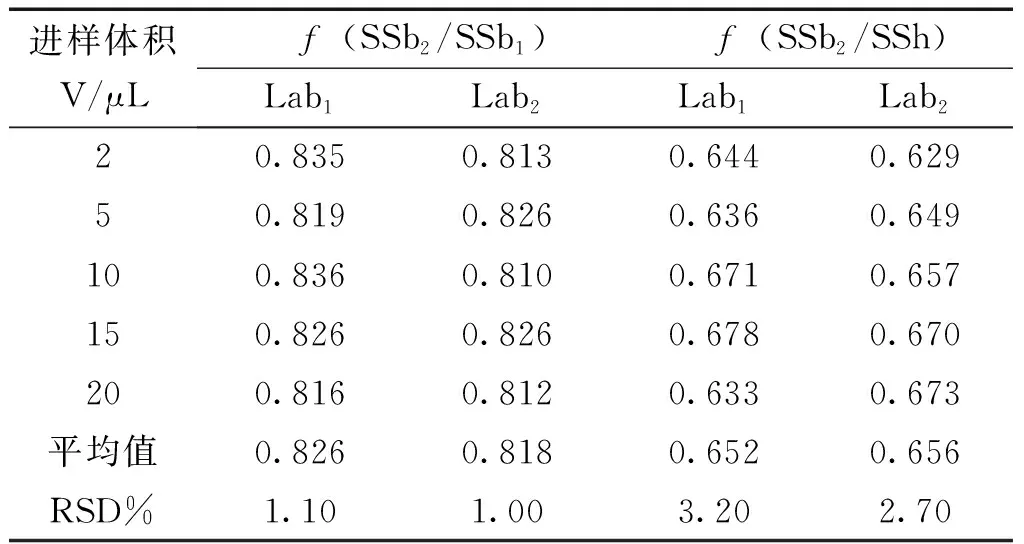
表4 不同实验室测得的相对校正因子
2.4 样品测定 取不同批次柴胡药渣皂苷富集物,每批3份,按照“2.1.3”项下方法制备供试品溶液,并依法测定,计算样品中各次生柴胡皂苷的含量,分别采用一评多测法和外标法计算柴胡皂苷h、b1的含量。结果见表5。
2.5 统计分析 采用统计学t检验,对表5中一评多测法和外标法得到的柴胡药渣中柴胡皂苷b1、h的含量进行比较,结果显示P值均大于0.05,表明两种方法测得的柴胡皂苷b1、h含量没有显著性差异。可见所建立的QAMS 法准确可行,可以替代外表法测定柴胡皂苷b1、h。

表5 两种方法测定不同批次柴胡药渣中柴胡皂苷的含量 (mg/g)
注:A外标一点法测定值 B一评多测计算法
3 讨论
针对于水提柴胡工艺的药渣中含有的柴胡皂苷,建立了如下的提取富集工艺:以含0.05%NaOH的50%乙醇-水溶液为提取溶媒,采用溶剂回流的方式进行提取;富集工艺采用大孔吸附树脂法[9],首先采用碱性水溶液进行洗脱除杂,然后用不同浓度的乙醇进行洗脱,收集高浓度的乙醇洗脱部位,浓缩,干燥,即得柴胡总皂苷。
对于检测波长的选择,通过对供试品和对照品溶液在不同波长下的图谱进行比较,发现254nm下,3种柴胡皂苷的吸收强度较大、图谱基线平稳,且分离度较好,故将测定波长确定为254nm。实验考察了不同的仪器、色谱柱,不同的实验条件下,柴胡皂苷b2对柴胡皂苷b1和h的相对校正因子的影响。结果显示,在上述不同的条件下相对校正因子重现性良好,RSD值均小于5%。为了检验所建立方法的准确性,采用外标法与一评多测法计算值进行反复验证,结果显示各待测成分实际含量与计算含量经过t-检验没有显著性差异。
不同于外标法,一评多测法能否成功应用的关键在于如何对测定对象的色谱峰进行准确定位[5]。本实验中,在所确定的色谱条件下,经过除杂、富集所得柴胡药渣皂苷的色谱峰相对较简单,干扰峰较少,通过3种柴胡皂苷间的相对保留时间即可实现目标成分的准确定位。
综上,本实验所建立的一评多测法测定柴胡药渣中总皂苷含量的方法具有较高的重现性、稳定性和可行度,有利于柴胡药渣总皂苷提取物的质量评价,证明了采用一评多测法测定同类化合物成分的可行性。
猜你喜欢
湖北农业科学(2021年8期)2021-05-11
家庭科学·新健康(2020年12期)2020-02-04
中成药(2019年12期)2020-01-04
中成药(2018年9期)2018-10-09
中成药(2018年4期)2018-04-26
中成药(2017年12期)2018-01-19
中成药(2017年9期)2017-12-19
中成药(2017年8期)2017-11-22
中成药(2017年6期)2017-06-13
食品与健康(2015年6期)2015-06-17
