
医疗器械单一审核程序中质量管理体系检查工作的介绍
2018-03-03 05:44:00温晶肖江宜王爱君国家食品药品监督管理总局食品药品审核查验中心北京市100061
中国医疗器械杂志 2018年1期
【作 者】温晶,肖江宜,王爱君国家食品药品监督管理总局食品药品审核查验中心,北京市,100061
1 MDSAP简介
为战略性加速国际医疗器械监管行为的协调统一,促进建立高效的医疗器械监管模式, 2011年10月,来自澳大利亚、巴西、加拿大、中国、欧盟、日本、美国和世界卫生组织(WHO)医疗器械监管机构的八方代表在渥太华召开会议,宣布成立国际医疗器械监管者论坛(International Medical Device Regulators Forum,简称IMDRF)。国家食品药品监督管理总局于2013年3月经国务院批准正式加入该组织。
医疗器械单一审核程序(Medical Device Single Audit Program,以下简称MDSAP)是IMDRF设置的工作项目之一。该项目旨在制定一套审核程序统一满足多个国家监管机构(Regulatory Authority)的审核要求,主要用于经评估获得认可的检查机构(Auditing Organization)[1-2]对医疗器械生产企业的质量管理体系进行的检查(以下简称体系检查)。通过该项程序,能够推动各国法规、检查标准的统一及检查成果的互认和共享,同时减轻了各国监管机构和生产企业的负担。该项目自2014年1月1日起开始实施试点,试点成员国为美国、日本、加拿大、巴西、澳大利亚,2017年1月开始在这5国正式实施。监管机构、检查机构及生产企业三者的关系见图1。
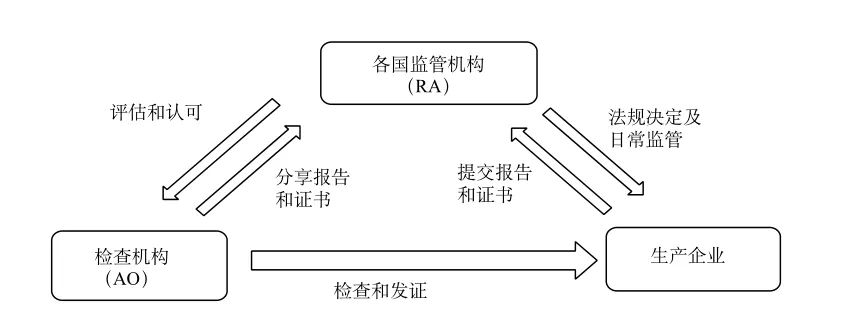
图1 监管机构、检查机构及生产企业关系图Fig.1 Relationship among RA, AO and manufacturers
2 体系检查的周期和类别
检查周期一般为3年。
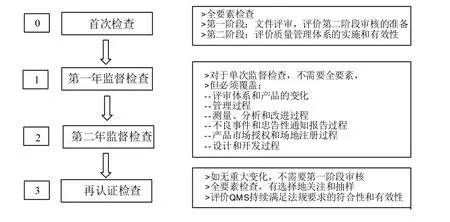
图2 体系检查三年周期图Fig.2 3-year QMS audit cycle
根据检查所处的周期不同,可以分为首次检查、监督检查和再认证检查[3]。具体见图2。根据检查的原因及实施主体不同,可以分为特殊检查、飞行检查及监管机构主导的检查等[4]。
3 体系检查的目的、依据及相关流程
3.1 体系检查的目的
(1)确定生产企业的质量管理体系是否符合ISO/IEC 13485的相关要求[5]。
(2)评价生产企业的质量管理体系是否包含了申请国法规中适用条款的要求以及相关要求是否得到有效实施。
3.2 体系检查的法规要求
质量管理体系标准:ISO/IEC 13485医疗器械—质量管理体系—用于法规的要求。
参与国相关法规的要求[6],具体内容见参考文献[6]。
3.3 体系检查的基本流程
体系检查的流程基本遵照ISO/IEC 17021-1中的相关规定,可分为资料审核、首次检查、监督检查及再认证检查四部分,大致内容参见ISO/IEC 17021-1:2015 “附录 E:第三方检查和认证过程”。
4 MDSAP检查任务的分类
在整个检查流程中,以首次检查为例,经过第一阶段检查后,检查机构会选定有能力的检查组在生产企业的现场进行第二阶段检查。该阶段主要以ISO/IEC 13485的审核为基础,兼顾各国的法规要求是否被融入质量管理体系并有效执行。
以ISO/IEC 13485:2016结合法规的检查为例,现场体系检查制定了非常详细的检查内容,主要包括管理过程,产品市场授权和设备注册,测量、分析和改进,医疗器械不良事件和忠告性通知报告,设计和开发,产品和服务控制以及采购共计7个部分,共涉及检查任务90项[7],具体情况见表1。

表1 体系检查的检查任务汇总表Tab.1 Summarized tasks of QMS audit
5 体系检查的结果
5.1 不符合项
不符合项的确定是依据GHTF/SG3/N19:2012《质量管理体系—医疗器械—为监管目的和信息交换而制定的不符合项分级系统》[8]中的要求执行的,具体情况见表2。
各类别所对应的分级之和为不符合项的最终等级,这种分级计划下的不符合的最终等级将是1到6之间的数字。然而“5”的等级被确定为最大值,因为这表示需要进行某种干预的足够高的风险,5级和6级之间的区别在分级系统中不被认为是有益的。 因此,如果达到6级,则最终等级记录为“5”。分级为1~3级为中低风险,4级和5级为风险程度较高的严重不符合项。
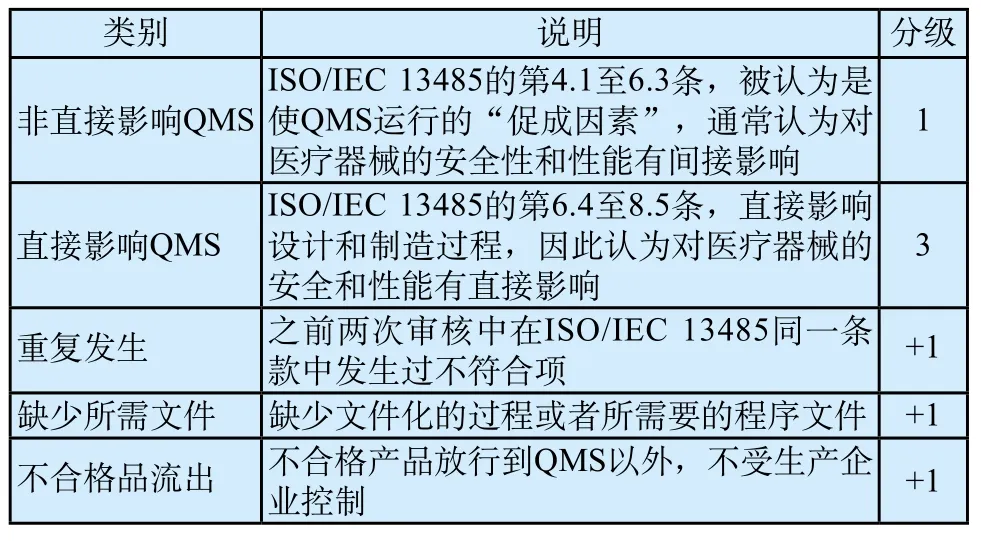
表2 不符合项分级系统Tab.2 Nonconformity grading system
5.2 检查报告[6,9]
MDSAP程序对检查报告制定了统一的模板,模板共分为19部分,包含了检查机构信息、检查场地信息、检查类别和标准、检查和认可范围、检查目的和检查时间、检查场地描述、关键供应商、以往检查情况、排除和不适用QMS要求的情况、第一阶段检查情况、检查发现(此部分是重点,需要按照检查任务详细表述,不符合项的内容需要注明详细信息并按照GHTF/SG3/N19:2012《质量管理体系-医疗器械-为监管目的和信息交换而制定的不符合项分级系统》中的要求进行分级)、与检查计划的重大偏差、检查阻碍(不配合)情况、以往不符合项的跟进情况、检查场地的主要变化、检查结论、附件(检查计划、不符合项分级表、不符合项报告包含对以往不符合项的检查情况)、检查组成员和报告起草人信息、报告批准等。
检查结果仅作为参考,各国监管机构将根据本国法律批准的流程和政策决定是否采用相关内容。
6 可以开展MDSAP检查的检查机构情况
根据MDSAP网站公布的相关内容[10],截止2017年5月15日,共有14家检查机构向MDSAP项目组提交了检查申请,11家机构获得授权可以开展MDSAP检查,目前仅有BSI Group America Inc.,Intertek Testing Services NA Inc.及TUV SUD America Inc.三家机构获得了认可,可以发放MDSAP认证证书,具体内容见参考文献[10]。
7 申请MDSAP的生产企业情况
截止2017年2月底,全球已经有262家企业申请了MDSAP检查,在我国境内的有4家。由于MDSAP仅针对第三方机构进行审核,不能将申请检查的生产企业名单向外公布,仅限于监管机构内部交流。如果想知道生产企业的相关信息,可以去已经通过审核的第三方机构官网上查询。
8 对我国检查机构的启示
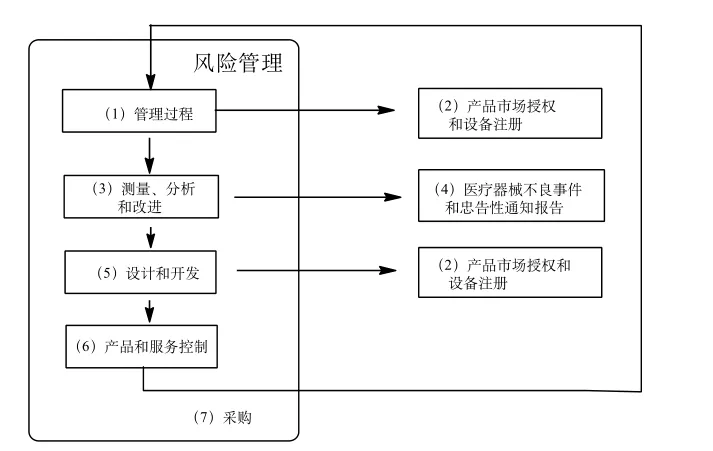
图3 风险管理与检查项目关系图Fig.3 Relationship between risk management and audit processes
8.1 制定细致的检查计划
以第二阶段现场检查为例,第三方机构制定了较为完整和详细的检查计划,计划中除列明了检查场所、地址、检查组人员、其他人员、检查范围、检查时长、检查标准和目的外,以表格的形式细致规划了每位检查员每个时间段的工作任务,让各方人员能够对每天的任务一目了然。由于之前经过了第一阶段审核,检查机构对生产企业的整体情况较为了解,检查组长对检查人员的能力也较为了解,因此制定检查计划的基础较为扎实,现场基本也能够按照该计划开展检查,取得了较好的效果。
8.2 对不符合项的证据描述要细致准确
MDSAP检查得出的各项不符合项的结论不能笼统或简要地说明理由,必须要有相关证据支持,最好列明企业自身的规定作为对比。如检查中“人员资质管理考核证据不充分,持续考核不完整”的结论需要提供如下证据:作为记录培训和证明能力的人员履历表,某位产品监管人员的表格并未更新,与培训文件QR-12-186-B/0中的相关规定不符。
8.3 关注检查项目之间的关联性
MDSAP7部分检查项目的具体任务之间经常有提示性链接,提示检查人员要考虑不同项目之间的关联性。一个项目的检查发现,经常对下一个项目的抽样环节起指导作用,例如在测量、分析和改进项目中发现的异常情况,可以在研发或者采购过程中作为抽样的指导依据。这就要求检查组必须做好前期计划分工和加强检查人员的现场交流,这就对检查组组长的整体规划能力及团队的整体合作能力提出了更高要求。
8.4 强化对风险管理的关注度
在检查全过程中,MDSAP非常关注质量管理体系各个项目是否贯彻了风险管理的相关理念。MDSAP 7部分检查项目与风险管理的关系见图3。检查人员善于以不良事件及抱怨为切入点,寻找其与设计开发和测量、分析改进之间的关系,研究判定相关风险点。
9 结语
作为医疗器械监管领域的最新成果,MDSAP程序已获得多个国家认可,检查流程和检查任务制定得较为详细,标准化和可操作性较好,相关内容对于我国检查机构制定检查计划、规范检查流程、突出检查重点、统一检查报告格式和内容等方面具有较强的参考价值。同时,考虑到目前一些地方检查机构通过政府购买服务的方式让第三方机构参与到医疗器械体系检查活动中来,MDSAP的相关内容也可以作为评价第三方检查机构检查能力和水平的重要参考。
[1] IMDRF MDSAP Working Group. IMDRF MDSAP WG/N3(ed2)– requirements for medical device auditing organizations for regulatory authority recognition[EB/OL] . 2016-03-24. https://www.fda.gov/downloads/MedicalDevices/InternationalPrograms/MDSAPPilot/UCM505689. pdf.
[2] IMDRF MDSAP Working Group. IMDRF MDSAP WG/N4 – competence and training requirements for auditing organizations[EB/OL]. 2013-12-09. https://www.fda.gov/downloads/MedicalDevices/InternationalPrograms/MDSAPPilot/UCM505243. pdf.
[3] ISO/IEC 17021-1:2015 – conformity assessment – requirements for bodies providing audit and certif i cation of management systems[S].
[4] IMDRF MDSAP Working Group. MDSAP AU P0002.004 –Audit Model[EB/OL]. 2017-01-06. https://www.fda.gov/downloads/MedicalDevices/InternationalPrograms/MDSAPPilot/UCM390382.pdf.
[5] ISO/IEC 13485:2016 –medical devices – quality management systems – requirements for regulatory purposes[S].
[6] IMDRF MDSAP Working Group. MDSAP AU F0019.1.005 –medical device regulatory audit report[EB/OL]. https://www.fda.gov/downloads/MedicalDevices/InternationalPrograms/MDSAPPilot/UCM387055.pdf.
[7] IMDRF MDSAP Working Group. MDSAP AU G0002.1.004 –revised 2017-04-17[EB/OL]. 2017-01-06. https://www.fda.gov/downloads/MedicalDevices/InternationalPrograms/MDSAPPilot/UCM390383.pdf.
[8] Study Group 3 of the Global Harmonization Task Force. GHTF/SG3/N19:2012 – nonconformity grading system for regulatory purpose and information exchange[EB/OL]. 2012-11-02. https://www.fda.gov/downloads/MedicalDevices/InternationalPrograms/MDSAPPilot/UCM468937.pdf.
[9] IMDRF MDSAP Working Group. MDSAP AU P0019.003– MDSAP Regulatory audit report policy[EB/OL]. 2016-08-15. https://www.fda.gov/downloads/MedicalDevices/InternationalPrograms/MDSAPPilot/UCM379903.pdf.
[10] IMDRF MDSAP Working Group. Auditing organization availability to conduct MDSAP audits[EB/OL]. 2017-05-15. https://www.fda.gov/downloads/MedicalDevices/InternationalPrograms/MDSAPPilot/UCM429978.pdf.
猜你喜欢
机械工业标准化与质量(2022年3期)2022-08-12 02:30:38
活力(2021年4期)2021-07-28 05:35:18
大众投资指南(2021年35期)2021-02-16 01:06:12
航天工业管理(2020年9期)2020-12-28 00:37:54
医疗装备(2020年10期)2020-06-13 01:34:36
质量安全与检验检测(2019年3期)2019-07-31 06:37:00
质量安全与检验检测(2018年6期)2018-12-28 06:23:46
中国医疗保险(2017年6期)2017-07-18 11:28:19
中国卫生(2016年5期)2016-11-12 13:25:50
中国卫生(2015年10期)2015-11-10 03:14:22
