
一步法合成甘草次酸甲酯及其结构研究
2018-03-02 09:18:38王海霞陈齐齐刘婷婷赵双双胡冬华
长春中医药大学学报 2018年1期
王海霞,陈齐齐,刘婷婷,赵双双,吴 勇*,胡冬华*
(1. 长春中医药大学药学院,长春 130117;2. 扬子江药业集团有限公司中药研究院,江苏 泰州 225321)
甘草又名美草、甜草、蜜草[1],大多分布在我国西北、华北、东北地区,是一种用途十分广泛的中药材[2]。据文献报道[3-6],国内外学者对其有效成分进行了大量深入的研究,包括提取、分离、结构改造、药理作用、临床应用等方面。其中对甘草酸和甘草次酸的研究最为广泛,甘草酸具有保肝解毒、抗病毒、抗炎以及增强免疫功能等作用,随着对其深入研究及临床应用表明,甘草酸对胃肠道有刺激作用,可引起假性醛固酮增多症[7],其表现是心脏和肌肉运作异常[8]。本研究以降低甘草酸的假性醛固酮增多症毒副作用为目的对甘草酸进行结构改造及合成条件优化,并在较低温度下,采用一步法合成甘草次酸的衍生物——甘草次酸甲酯,并对其合成条件进行优化。
1 仪器与材料
78-1型磁力加热搅拌器,AL 204分析天平,SHZCB型循环水式真空泵,WFH-203三用紫外线分析仪,INOVA-500核磁共振仪,X-5精密显微熔点测定仪,ZK-82B型电热真空干燥箱,TU-1810型紫外-可见分光光度计,YLE-2000恒温水浴锅,瑞利红外光谱仪。甘草酸98%,甘草次酸97%,甲醇,乙醇,盐酸,三氯甲烷,石油醚,甲苯,乙酸乙酯,浓硫酸,苯,乙酸,溴化钾,以上试剂均为分析纯。
2 方法与结果
2.1 甘草次酸甲酯的合成 称取甘草酸1.0 g置于50 mL烧杯中,加入10% HCl 10 mL、甲醇5 mL,室温搅拌12 h后转移至反应釜中,于烘箱90 ℃下反应48 h。反应结束后,取出,冷却至室温,过滤,将所得沉淀用水多次洗涤至无醇味,置50 ℃烘箱中干燥24 h。将所得干燥物用80%乙醇进行反复重结晶即得甘草次酸甲酯。
2.2 合成条件优化
2.2.1 正交因素、水平的确定 根据文献报道[9-11]影响甘草次酸甲酯合成主要的因素有反应时间、反应温度以及催化剂种类、浓度。本试验考察了反应时间、反应温度以及盐酸的浓度对合成产率的影响,采用L9(34)正交实验表。因素水平见表1。

表1 试验因素水平表
2.2.2 正交试验结果与分析 采用正交试验方法,以甘草次酸甲酯的产率为考察指标,考察反应时间、反应温度和盐酸浓度3个因素对合成产率的影响,结果见表2、表3。
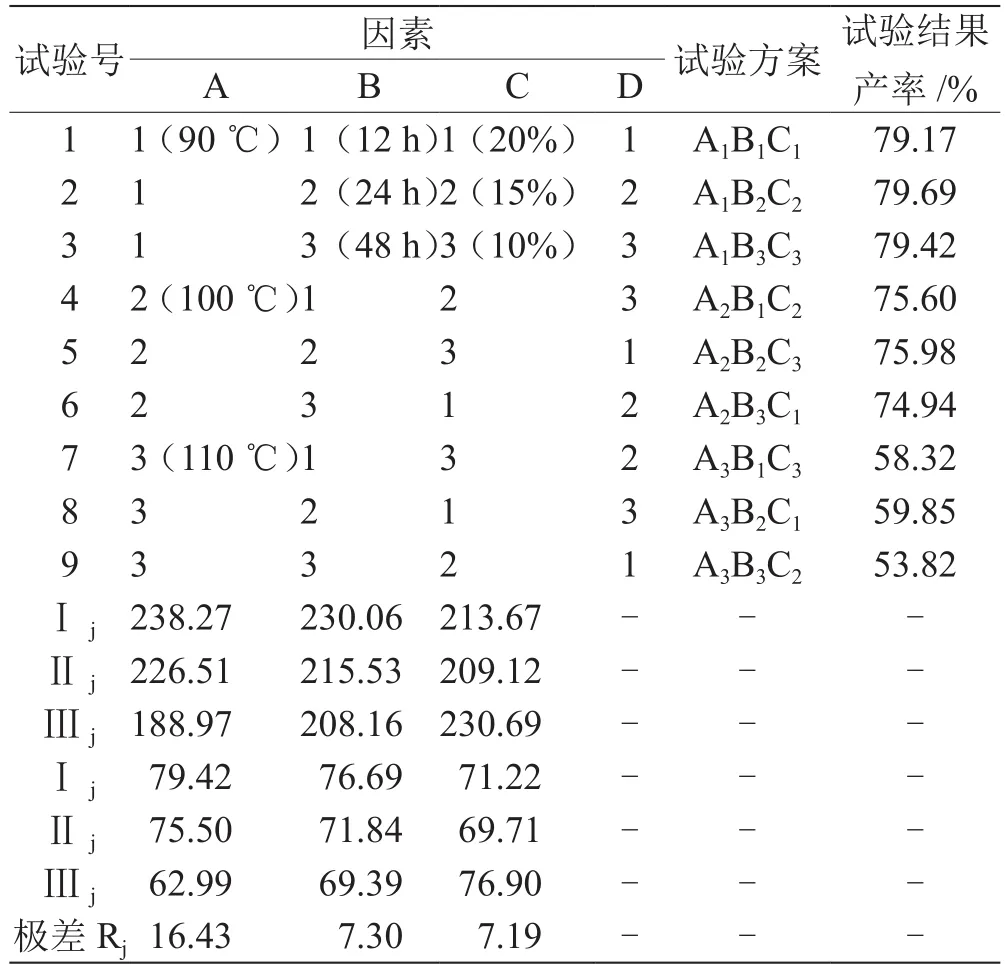
表2 正交试验结果与直观分析L9(34)
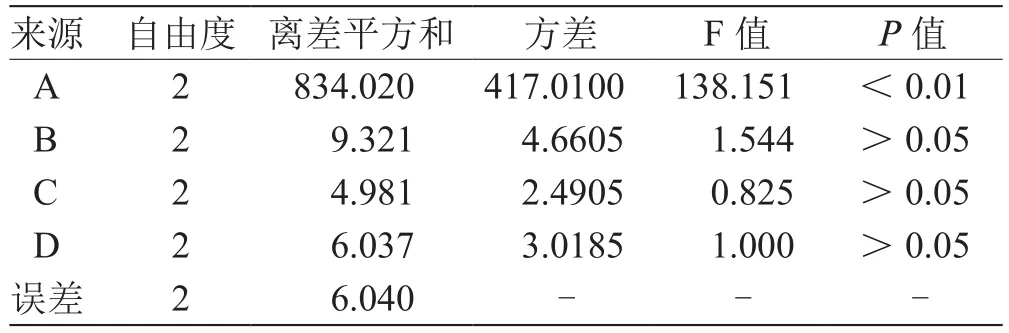
表3 方差分析表L9(34)
由方差分析表可知,各因素对合成甘草次酸甲酯反应的影响大小顺序为,A>B>C,即反应温度>反应时间>盐酸浓度。由均值分析可知,最佳合成条件为A1B1C3,即反应温度90 ℃、反应时间12 h、盐酸浓度(质量分数)10 %。本实验方法一步合成目标产物,合成条件更为温和,产物纯度高,合成效率好。
2.3 甘草次酸甲酯结构研究
2.3.1 物理性质 合成产物甘草次酸甲酯为白色粉末状晶体,于显微镜下观察为细小的白色针状结晶。通过溶解实验得知其易溶于氯仿,溶于乙醇、甲醇等,难溶于水。通过熔点测定仪测得其熔点为246~250 ℃(文献值250~251 ℃)[8]。
2.3.2 薄层层析鉴定 考察了不同展开剂系统,选用石油醚-甲苯-乙酸乙酯-乙酸进行展开。样品为2 mg/mL的三氯甲烷溶液,吸附剂为硅胶G;展开剂:石油醚 : 甲苯 : 乙酸乙酯 : 乙酸 = 5 : 10 : 3.5 : 0.28;显色剂:30 %硫酸溶液;置于365 nm紫外光灯下检视,结果:甘草次酸甲酯为甘草次酸的衍生物,其结构与甘草次酸不同之处在于甲氧基和羟基,故两者极性有差异。合成产物(标记2)的Rf值大于甘草次酸(标记1)的Rf值。由此得知该合成产物的极性小于甘草次酸,符合化合物发生酯化后的特征规律。
2.3.3 紫外-可见吸收光谱分析 取经纯化产物0.5 mg溶于100 mL 80 %乙醇中,以80 %乙醇为空白对照,置于TU-1810紫外-可见分光光度计中进行光谱扫描,结果合成产物甘草次酸甲酯有2个特征吸收峰,结果见表4。

表4 甘草次酸甲酯紫外-可见吸收光谱扫描结果
2.3.4 红外吸收光谱分析 将经纯化的产物置于烘箱中干燥,取1.5 mg,与200目干燥的KBr粉末(光谱纯)约200 mg,置于玛瑙乳钵中,用研杵研细并混合均匀,压成直径约13 mm、厚度约1 mm的透明KBr样品片,利用红外分光光度计进行扫描。结果可知,νas(-COOCH3)为1 726.59 cm-1,是酯类化合物的第一特征峰;νas(-C-O-C-)为 1 217.26 cm-1、1 156.14 cm-1,是酯的第二特征峰;νas(O=C<)为1 658.18 cm-1;νas(-OH)为 3 428.66 cm-1;νas(=C-H)为 994.23 cm-1,νas(-CH3)为 2 947.62 cm-1、1 464.20 cm-1、1 386.76 cm-1,这些特征吸收峰与甘草次酸甲酯结构都十分吻合,可以初步确定该合成产物为甘草次酸甲酯。
2.3.5 核磁共振波谱 核磁共振波谱在化合物结构确定方面具有重要作用,应用广泛,测定结果可靠。本研究采用核磁共振波谱检测,合成产物检测结果:
1H-NMR(600MHz,CDCl3) d:7.25(CDCl3,5.64(s,1H,12-H),3.71(s,3H,-COOCH3),3.22(m,1H,3-H),2.79(m,1H,18-H),2.23(s,1H,9-H),1.50~1.01(m,6H),1.70~1.58(s,3H,27-H),1.20~ 1.14(m,1H),1.19(s,3H,30-H),1.13(s,3H,25-H),1.16(s,3H,26-H),1.07~0.99(m,2H,1-H),1.02(s,3H,23-H),0.81(s,3H,CH3),0.80(s,3H,CH3),0.74~0.60(m,1H,5-H)。以上数据与文献[9]值相符,表明该合成产物为甘草次酸甲酯。
3 讨论
甘草酸属于五环三萜类化合物,其结构中的羰基和羧基是甘草酸发挥药效时引起不良反应的主要基团,因此本研究针对其中的羧基和葡萄糖醛酸进行结构修饰,合成目标化合物甘草次酸甲酯。文献中关于甘草次酸甲酯的合成主要有两种方法,一种是先在高温高压条件下以酸为催化剂将甘草酸进行水解,生成甘草次酸;然后在酸存在的条件下甘草次酸与甲醇进一步进行酯化反应[9],另一种是采用克莱门森还原的方法[12]。本研究采用一步水热法合成甘草次酸甲酯,省去了甘草酸单独进行水解的步骤,实验中所用的酸既可以催化甘草酸水解,又可以作为酯化反应的催化剂,节省了原料,避免了中间过程的物料损失;提高了产品的产率;产物的纯化通过反复重结晶即可完成,操作简单,产品纯度较高。
本研究结果证明,甘草次酸甲酯具备甘草酸的基本药用价值,比如治疗肝损伤、抗溃疡、抗炎等活性。但是甘草次酸甲酯比甘草酸更具有优越性,其假性醛固酮增多症毒副作用大大降低[13],并且脂溶性更好,更有利于人体吸收。
[1]国家药典委员会.中华人民共和国药典[M].北京:化学工业出版社, 2015.
[2]李海华,青梅.甘草的研究进展[J].内蒙古医科大学学报,2015(2):199-204.
[3]李玉山.甘草酸提取纯化工艺的研究进展[J].化学试剂,2016, 38(5):428-432.
[4]金京丽,尹明浩.浅述甘草古今运用[J].吉林中医药,2006, 26(4):52-53.
[5]张利.甘草的药理作用及现代研究进展[J].中医临床研究,2014, 6(10):147-148.
[6]周彪,万传星.甘草地上部分化学成分研究[J].中草药,2016, 47(1):21-25.
[7]吴楠.甘草次酸衍生物药效学筛选及相关机制的探讨[D].天津:天津医科大学, 2006.
[8]覃瑶,王欣.甘草次酸衍生物的抗炎活性及水钠潴留作用的研究[J].华西药学杂志, 2013, 28(4):338-340.
[9]刘文丛,罗云清.水热法合成甘草次酸甲酯的研究[J].东北师大学报(自然科学版), 2007, 39(4):154-156.
[10]陈兰,李贵香.系列甘草次酸衍生物的合成与波谱测定[J].中国药房, 2014(3):249-252.
[11]韩亚男,程新宇,侯俊玲,等.大孔树脂纯化甘草地上部分总黄酮的工艺[J].吉林中医药, 2014, 34(1):82-86.
[12]雍建平.阿吉艾克拜尔·艾萨.甘草次酸/11-脱氧甘草次酸甲酯的合成[J].时珍国医国药, 2011, 22(11):2618-2619.
[13]木合布力·阿布力孜,马淑燕,闵杰.甘草次酸的结构修饰研究进展[J].新疆医科大学学报, 2007, 30(2):185-187.
猜你喜欢
云南化工(2020年11期)2021-01-14 00:50:52
中成药(2018年2期)2018-05-09 07:20:08
中成药(2017年3期)2017-05-17 06:08:48
中国卫生标准管理(2015年4期)2016-01-14 05:16:45
山东医药(2015年16期)2016-01-12 00:40:04
西南医科大学学报(2016年4期)2016-01-03 01:26:29
化工进展(2015年3期)2015-11-11 09:07:41
中国当代医药(2015年33期)2015-03-01 02:09:17
中国当代医药(2015年10期)2015-03-01 02:02:39
海军医学杂志(2015年2期)2015-02-27 13:47:35
