
群体构成方式对大豆百粒重全基因组选择预测准确度的影响
2018-01-18 01:29:28马岩松刘章雄文自翔魏淑红杨春明王会才杨春燕卢为国张万海0吴纪安胡国华栾晓燕付亚书王曙明韩天富张孟臣苑保军JochenREIF李文滨王德春邱丽娟
作物学报 2018年1期
马岩松 刘章雄 文自翔 魏淑红 杨春明 王会才 杨春燕 卢为国 徐 冉 张万海0 吴纪安 胡国华 栾晓燕 付亚书 郭 泰 王曙明 韩天富 张孟臣 张 磊 苑保军 郭 勇 Jochen C. REIF 江 勇 李文滨 王德春 邱丽娟,*
群体构成方式对大豆百粒重全基因组选择预测准确度的影响
马岩松1,2,13刘章雄1文自翔3魏淑红4杨春明5王会才6杨春燕7卢为国8徐 冉9张万海10吴纪安11胡国华12栾晓燕13付亚书14郭 泰15王曙明5韩天富1张孟臣7张 磊16苑保军17郭 勇1Jochen C. REIF18江 勇18李文滨2王德春3邱丽娟1,*
1中国农业科学院作物科学研究所 / 国家农作物基因资源与遗传改良重大科学工程 / 农业部作物种质资源与生物技术重点开放实验室, 北京 100081;2东北农业大学农学院, 黑龙江哈尔滨150030;3Department of Plant, Soil and Microbial Sciences, Michigan State University, East Lansing, MI 48824, USA;4黑龙江省农业科学院育种研究所, 黑龙江哈尔滨 150081;5吉林省农业科学院大豆研究所, 吉林长春 130033;6内蒙古赤峰市农科所, 内蒙古赤峰 024031;7河北省农业科学院粮油作物研究所, 河北石家庄 050031;8河南省农业科学院经济作物研究所, 河南郑州 450002;9山东省农业科学院作物研究所, 山东济南 250010;10内蒙古呼伦贝尔市农科所, 内蒙古呼伦贝尔 021000;11黑龙江省农业科学院黑河分院, 黑龙江黑河 164300;12黑龙江省农垦科研育种中心, 黑龙江哈尔滨 150090;13黑龙江省农业科学院大豆研究所, 黑龙江哈尔滨 150086;14黑龙江省农业科学院绥化分院, 黑龙江绥化 152052;15黑龙江省农业科学院佳木斯分院, 黑龙江佳木斯 154007;16安徽省农业科学院作物研究所, 安徽合肥 230031;17河南省周口市农业科学院, 河南周口 466001;18Department of Breeding Research, Leibniz Institute of Plant Genetics and Crop Plant Research (IPK), Gatersleben 06466, Germany
百粒重是大豆产量的重要构成因子, 在一定条件下与产量呈显著正相关。百粒重是一个复杂的数量性状, 用传统的育种方法其遗传增益不明显。本研究对280份大豆品种进行了多年多点田间鉴定, 通过混合线性模型预测获得品种百粒重的最佳线性无偏预测值。同时利用分布在大豆全基因组的5361个SNP标记鉴定参试品种基因型, 结合随机回归最佳线性无偏预测模型和交互验证方法, 探讨了群体构成方式对大豆百粒重的全基因组选择预测准确度的影响。结果表明, 大豆百粒重的全基因组选择预测准确度变化范围为–0.15~ +0.75; 群体构成方式对百粒重的预测准确度影响明显; 亚群内的预测准确度(+0.24~ +0.75)高于亚群间(-0.15~ +0.29); 当群体间遗传距离由0.1566增加到0.2201时, 预测准确度下降27.87%; 相比随机构建的训练群体, 基于群体遗传结构构建的训练群体能将百粒重的预测准确度提高2.34%。本研究明确了大豆百粒重的全基因组选择预测准确度, 阐明了群体结构对大豆百粒重的全基因组选择预测准确度的影响, 为大豆分子育种提供了新的思路和方法。
大豆; 百粒重; 全基因组选择; 预测准确度; 遗传结构
大豆是人类植物蛋白和脂肪的重要来源, 在日常膳食中占重要地位[1]。随着生活水平的提高和膳食结构的调整, 我国对大豆的需求量与日俱增, 而我国大豆单位面积产量与世界平均水平差距仍较大,联合国粮食及农业组织的数据(http://www.fao.org/ faostat/zh/#data/QC)表明, 在1961—2014年间我国大豆与美国、阿根廷和巴西的大豆单产平均差距为41%~66%。因此, 如何有效提高我国大豆的单产水平是大豆育种中亟待解决的重大课题。
百粒重是大豆产量的重要构成因子, 在一定条件下大豆百粒重与大豆产量呈显著正相关[2-3]。大豆百粒重相关QTL研究已取得显著进展, 前人利用分离群体、自然群体、重组自交系和染色体片段代换系等群体, 结合复合区间作图法、关联分析和Meta分析等方法定位了大量与大豆百粒重相关的QTL[4-7]。截止到2015年, SoyBase数据库收录的与大豆粒重相关QTL共计230个(http://soybase.org/ search/index.php?search= true&result=qtl &qtl=Seed+ weight)。这些相关研究为开发分子标记并用于育种创造了条件。然而, 标记辅助选择应用于百粒重等由微效多基因控制的复杂数量性状具有一定局限性, 全基因组选择则为复杂性状遗传改良提供了新方法[8-10]。
全基因组选择(GS)由Meuwissen等[11]首先提出, 是标记辅助选择(MAS)的一种新方法。该方法利用分布在染色体的高密度分子标记估算候选个体的基因组估计育种值(GEBV), 并以此作为候选个体选择的标准。与传统的标记辅助选择(MAS)和基因组关联分析(GWAS)相比, 全基因组选择无需选择标记, 而将所有标记用于估计育种值, 以提高对微效多基因控制复杂性状的选择效率[11]。近年来, 全基因组选择已被广泛应用于玉米[12-13]、小麦[14-15]、大豆[16-17]、水稻[18]、大麦[19]、黑麦[20]、向日葵[21]等作物和桉树[22]等林木育种研究。全基因组选择的关键是通过交互验证方法构建基于分子标记选择目标性状的预测模型。全基因组选择首先利用同时具有表型数据和基因型数据的训练群体(TS), 建立目标性状全基因组选择的预测模型, 再利用预测模型分析验证群体(VS)的基因型并预测其表现型, 通过表型鉴定验证预测结果的准确度。预测准确度通常由基因组估计育种值(GEBV)与表型值的Pearson相关系数表示。全基因组选择预测准确度的影响因素很多, 主要包括群体连锁不平衡程度、标记类型和数量、目标性状遗传力、训练群体与验证群体的关系、预测模型的选择等[12-24]。
大豆的基因组序列数据的公布[25]及重测序[26-28]为全基因组选择提供了丰富的标记信息, 50k大豆芯片的开发与利用[29]为大豆全基因组选择实践提供了技术支持。然而, 关于大豆全基因组选择影响因子方面的报道还较少。Shu等[16]利用基于内含子序列开发的79个SCAR标记对288个大豆品种百粒重进行全基因组选择分析, 使用随机回归最佳线性无偏预测模型(RRBLUP)和贝叶斯线性回归模型(Bayesian linear regression)的预测准确度最高值分别为0.854和0.904。Bao等[17]用282个品种的1536个SNP标记预测大豆抗胞囊线虫病的全基因组选择准确度的范围为0.59~0.67, 随着标记数量的降低其预测准确度呈下降趋势。然而, 有关群体结构对大豆全基因组选择预测准确度影响相关研究尚未见报道。
本研究以280份大豆品种组成的自然群体为材料, 利用分布在大豆全基因组的5361个SNP标记和多年多点表型估算百粒重数据, 分析供试群体的遗传结构、训练群体与验证群体的构成方式等对大豆百粒重的全基因组选择预测准确度的影响, 为大豆产量相关性状的全基因组选择育种提供理论依据。
1 材料与方法
1.1 供试群体的构成及表型鉴定
280份大豆育成品种(系)构成的自然群体包括北方春大豆240份, 黄淮夏大豆39份, 引进国外种质1份(见附表1)。
2008—2010年表型鉴定试验地点为黑龙江、吉林、内蒙古、河北、河南和山东。采用随机区组田间设计, 3次重复, 4行区, 行长5 m。2011—2012年表型鉴定试验地点为黑龙江、吉林、内蒙古、河北、河南、山东和安徽。采用随机区组田间设计, 2次重复, 3个行区, 行长3 m。均采用试验地常规大豆种植管理方式。收获时每个小区随机选择中间长势均匀的10个单株参照邱丽娟等[30]编著的《大豆种质资源描述规范和数据标准(2006)》调查百粒重数据。
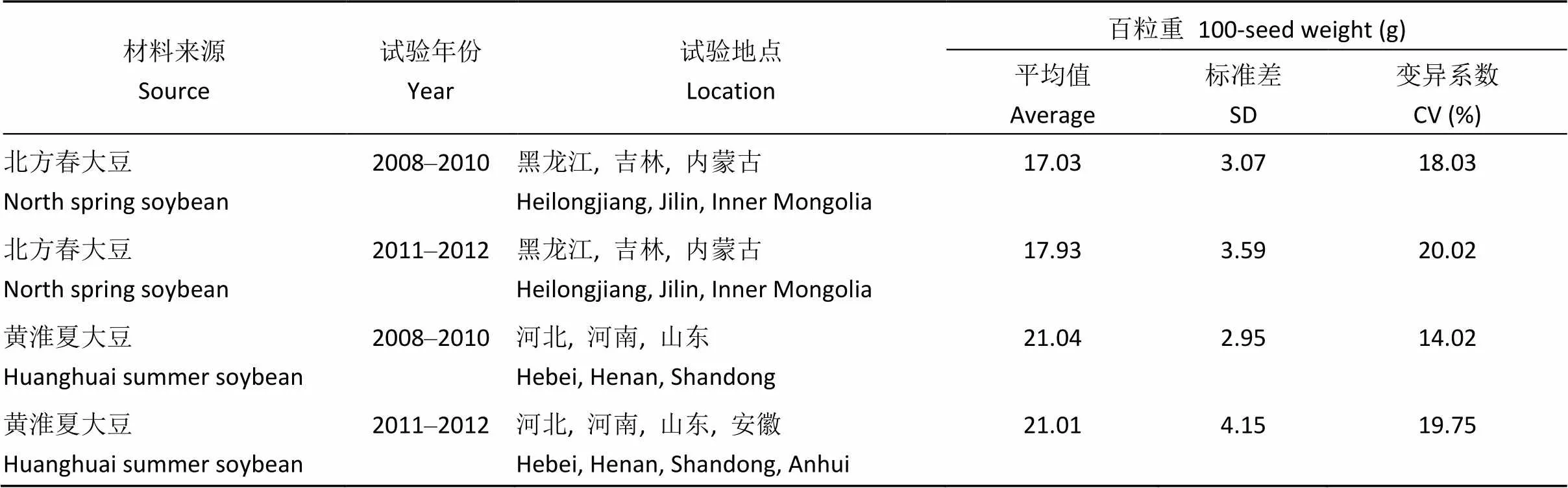
表1 不同鉴定年份及试验地点间百粒重变化
1.2 供试群体目标性状最佳线性无偏预测值和遗传力的计算
针对本研究的表型性状数据为非均衡数据, 采用混合线性模型,Y =+G+Y+L+。其中Y表示第个品种在第年的地点的目标性状观察值,代表目标性状群体平均数,G、Y和L分别代表基因型(品种)效应、年份效应和地点效应,代表随机误差。将基因型(品种)、年份和地点作为随机效应。计算参试品种目标性状的最佳线性无偏预测值(BLUP), 以此作为目标性状的基因组选择表型数据。采用费尔等[31]的分析方法计算数量性状遗传力。
22 / (2 + σ2/2/2/2/)
式中,2表示广义遗传力,2表示基因型方差,2表示基因型与年份互作方差,2表示基因型与地点互作方差,2表示基因型、年份和地点互作方差,表示试验年份,表示试验地点,表示重复次数。
1.3 供试群体的基因型鉴定
利用Illumina SoySNP 6k iSelect BeadChip大豆芯片完成供试群体的全基因组扫描。该芯片由分布于大豆20条染色体上的5361个SNP组成。这些SNP标记是根据已发表大豆重要农艺性状QTL定位区间, 选自于Song等[29]构建的Illumina SoySNP50k iSelect BeadChip。利用GenomeStudio程序检测获得供试群体的基因型数据。删除缺失数据超过5%的标记, 筛选出5354个SNP标记用于进一步分析。
1.4 群体遗传结构分析
采用基于贝叶斯理论的马尔科夫链蒙特卡罗方法(MCMC)及Structure 2.2软件分析供试群体遗传结构。亚群数量(值)设定范围为1~10, Burn-in次数和MCMC重复次数均为10 000。依据Evanno等[32]D方法确定值。
利用主成分分析方法(principle component analysis, PCA)结合TASSEL 5.0软件[33]分析供试群体的基因型。分别以第一主成分和第二主成分为坐标轴, 绘制供试群体散点图。
1.5 全基因组选择及预测准确度计算
采用随机回归最佳线性无偏预测模型(random regression best linear unbiased prediction, RR-BLUP)和5倍交互验证方法研究大豆百粒重全基因组选择。RR-BLUP模型为++e, 式中表示供试材料目标性状的最佳线性无偏预测值向量,表示群体平均值,表示标记的加性效应,= (X)表示´维基因型矩阵,表示残差项。模型中, 假设标记效应和残差项符合各自的随机分布, 即~(0,Iσ2α)和~(0,Iσ2), 其中I和I表示单位矩阵, 单位矩阵的维数分别为2=σ2/p和2=σ2/l,2和2分别表示基因型方差和误差方差,和分别表示标记数量和试验地点数量。
采用5倍交互验证方法, 首先将供试群体随机分成5份, 随机选择其中4份构成训练群体(training subset, TS), 剩余的一份为验证群体(validation subset, VS), 然后在训练群体中利用表型数据和基因型数据建立大豆全基因组选择模型, 最后, 在验证群体中利用基因型数据和预测模型估算基因组估计育种值(genomic estimated breeding value, GEBV)。全基因组选择预测准确度GSMP/, 其中MP表示验证群体中基因组估计育种值与实际观察值的相关系数,表示遗传力的平方根, 重复以上过程500次以消除取样误差。
1.6 训练群体及验证群体的构成方式
在供试群体中采用五倍交互验证方式估算供试群体总体的百粒重的全基因组选择预测准确度; 在群体遗传结构分析的基础上, 将供试群体分成若干亚群, 在每个亚群中利用相同方法估算百粒重的全基因组选择预测准确度; 分别以每个亚群为训练群体, 其他亚群为验证群体, 估算不同亚群间百粒重的全基因组选择预测准确度; 分别以其中一个亚群为验证群体, 其他亚群为训练群体, 估算不同亚群间百粒重的全基因组选择预测准确度; 将每个亚群随机分成5个部分, 随机选择每个亚群的任意4个部分组成训练群体, 每个亚群的剩余的一个部分为验证群体, 估算基于群体结构的训练群体对百粒重全基因组选择预测准确度的影响。
1.7 数据分析软件及方法
通过R语言的“lme4”数据包计算供试群体目标性状最佳线性无偏预测值和遗传力, 利用Structure 2.2软件分析群体遗传结构, 利用Tassel 5.0软件分析基因型主成分, 由R语言的“rrBLUP”数据包完成RRBLUP模型及5倍交互验证。
2 结果与分析
2.1 供试群体表型性状最佳线性无偏预测及遗传力
利用混合线性模型获得280份大豆品种的百粒重最佳线性无偏预测值, 供试品种的百粒重变化范围为13.39~23.72 g, 平均18.97 g, 变异系数为7.80%。其中北方春大豆百粒重变化范围为13.39~ 23.72 g, 平均18.92 g, 变异系数为7.45%; 黄淮夏大豆百粒重变化范围为16.15~23.49 g, 平均19.29 g, 变异系数为9.59% (表2)。与北方春大豆相比, 黄淮夏大豆的平均百粒重呈上升趋势但差异不显著(测验)。
方差分析表明, 品种间的百粒重差异均达到极显著水平。此外, 百粒重在年份间、试验地点间、年份与品种互作以及品种、地点和年份间互作均达到极显著水平。地点与品种互作间差异不显著。利用方差分析估算的百粒重遗传力较高为0.92 (表3)。

表2 不同群体间百粒重最佳线性无偏预测描述性分析
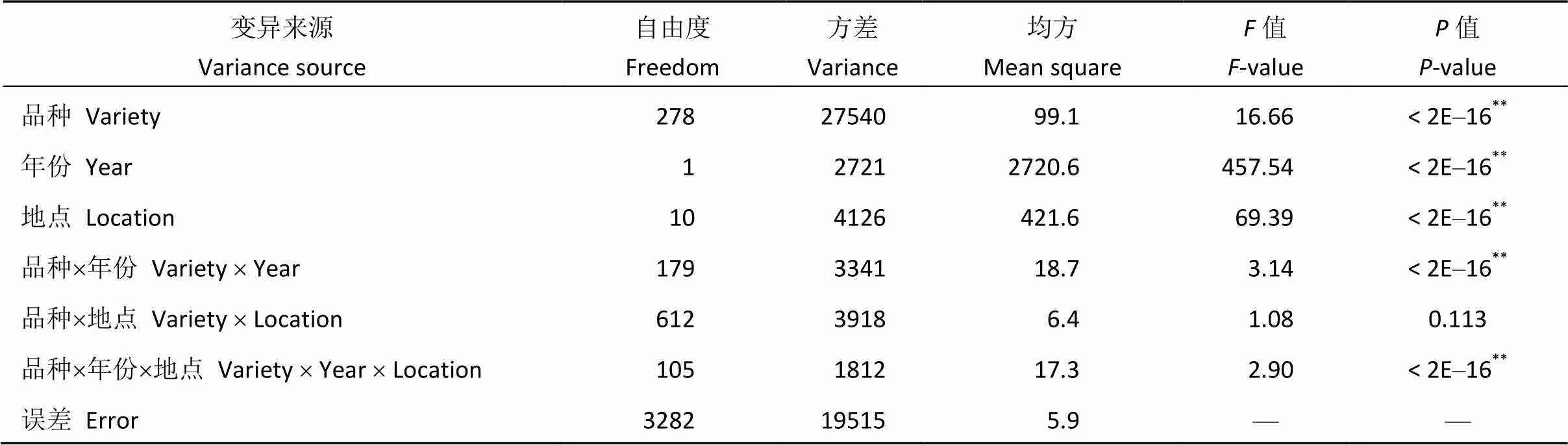
表3 供试品种百粒重方差分析及遗传力
**表示在0.01水平上差异显著。**means significant difference at the 0.01 level.
2.2 供试群体的遗传结构
利用Structure软件计算不同值下D的变化趋势。当= 3时,D最大, 并且随着值的增减,D显著降低(图1)。表明供试的280份大豆品种可被分成3个亚群。其中第I和第II亚群分别由133份和88份品种组成, 全部为北方春大豆, 第III亚群为混合亚群, 包括39份黄淮夏大豆和20份北方春大豆。不同亚群间的遗传分化指数, 亚群内期望杂合度以及亚群间遗传距离见附表2。
进一步分析不同亚群中品种来源发现, 第I亚群品种主要来自黑龙江省, 有115份, 占参试品种总数的86.46%, 其中吉林、内蒙古、辽宁品种分别为13份、3份和1份, 还有1份国外品种。第II亚群以吉林品种为主, 为75份, 占比85.23%, 黑龙江、辽宁和山西品种分别为9份、3份和1份。第III亚群的品种来源较广, 20份北方春大豆来源于辽宁、山西、吉林、北京、内蒙古、河北等7个省(市、自治区)。39份黄淮夏品种则来源于北京(11份)、山东(9份)、河南(6份)、河北(6份)、江苏(3份)、安徽(2份)、山西(2份) 7个省(市)。
利用5354个SNP基因型数据对280份大豆品种进行主成分分析。前2个主成分累计解释总变异的15.43%。分别以第一主成分与第二主成分为坐标轴, 绘制280份大豆品种散点图, 在第一主成分将第III亚群与第I亚群和第II亚群分成两部分, 在第二主成分, 第I亚群与第II亚群被分成两部分, 日本品种十胜长叶分到了第I亚群(图2)。
2.3 群体构成方式对全基因组选择预测准确度的影响
2.3.1 供试群体及不同亚群内目标性状预测准确度比较 在第I亚群内随机选择130份大豆品种, 第II亚群随机选择85份大豆品种, 第III亚群内随机选择55份大豆品种作为每个亚群的抽样群体。在每个抽样群体中以5倍交互验证方法估算每个亚群的百粒重的全基因组选择预测准确度。为消除取样误差, 以上过程均重复500次。通过比较发现, 百粒重的预测准确性在不同群体间差异明显, 以第III亚群最高为0.75, 第I亚群最低为0.24, 而总体和第II亚群分别为0.54和0.51 (表4和图3)。
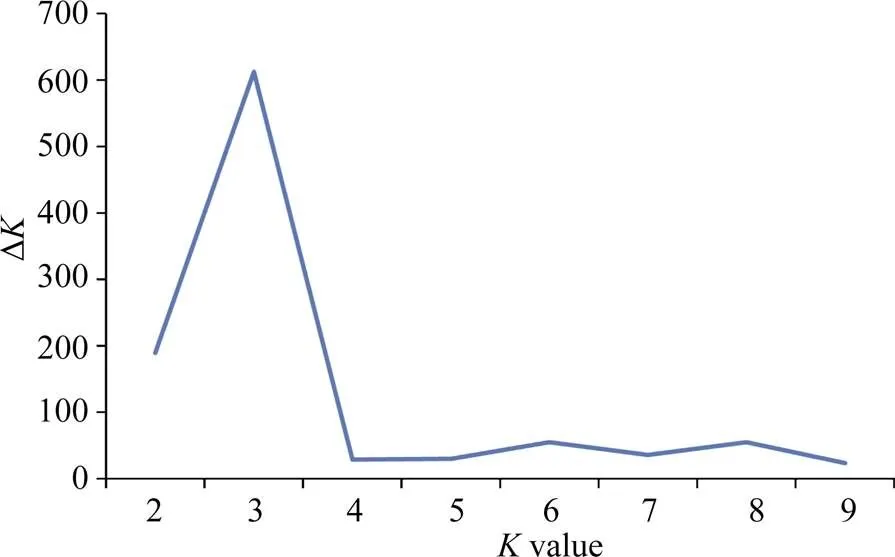
图1 不同K值下DK变化趋势

图2 280份大豆品种主成分分析散点图

表4 不同群体构成方式间百粒重预测准确度比较
C1、C2、C3分别表示第一亚群、第二亚群和第三亚群。
C1, C2, and C3 mean the first, the second, and the third subset, respectively.
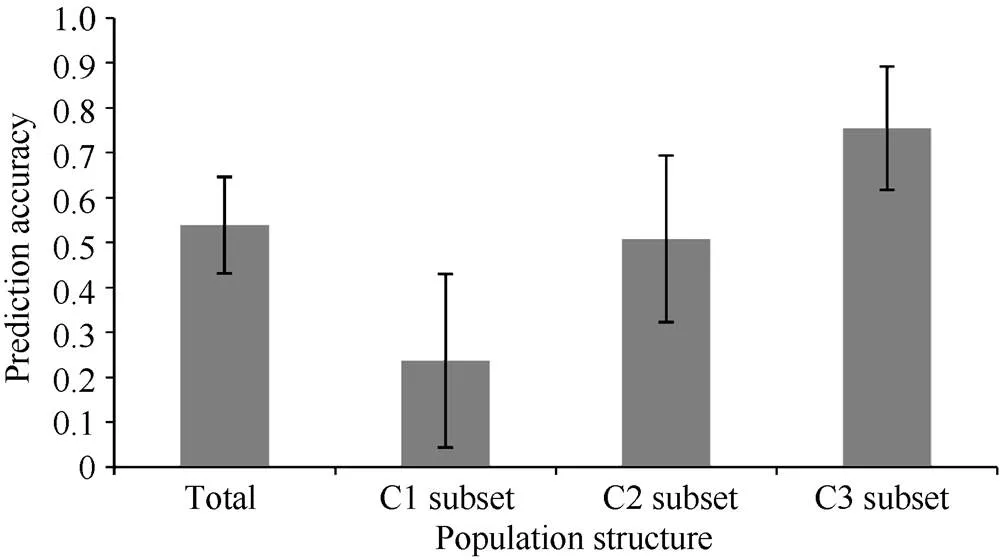
图3 不同亚群间百粒重预测准确度平均值及标准差柱状图
2.3.2 随机取样与基于群体结构取样构成的群体间预测准确度比较 为了比较群体结构对百粒重全基因组选择预测准确度的影响, 分别将3个亚群的抽样群体随机分成数量相等的5个部分, 在每个亚群中任意选择4个部分构成训练群体, 同时将每个群体的剩余的部分构成验证群体, 建立了基于群体结构的训练群体和验证群体, 估算目标性状的全基因组选择预测准确度, 重复500次以消除取样误差。利用2.3.1中总体的5倍交互验证方法估算百粒重的预测准确度为对照。不同方法构建的训练群体和验证群体目标性状预测准确度结果表明, 基于群体结构构建的训练群体和验证群体百粒重的预测准确度略高于随机取样构建群体间百粒重的预测准确度, 但差异经测验未达到显著水平(图4)。
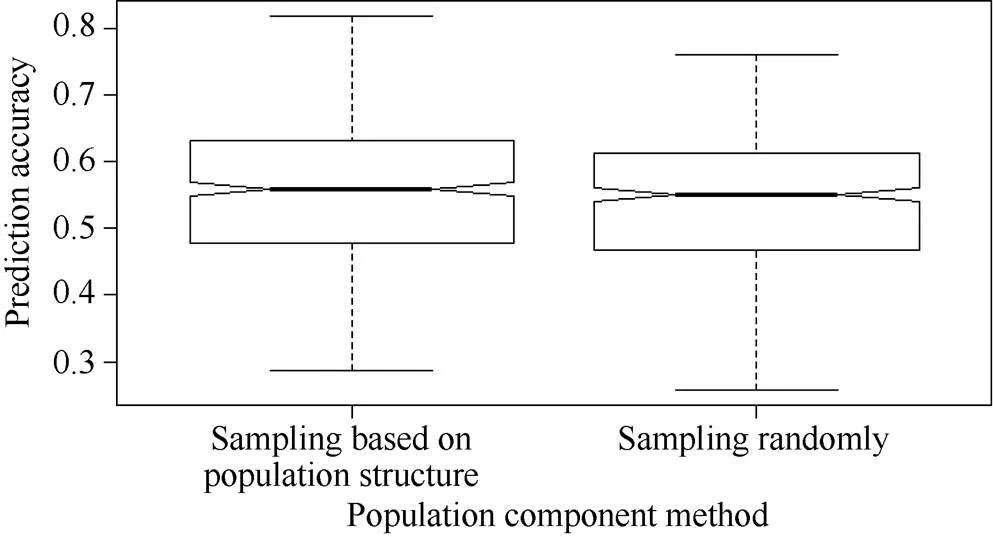
图4 不同方法构建的训练群体和验证群体百粒重的预测准确度箱线图
2.3.3 不同亚群间目标性状预测准确度比较 分别以每个亚群为训练群体, 预测另外2个亚群的百粒重基因组估计育种值, 并估算预测准确度(表3)。亚群内相比, 不同亚群间百粒重的预测准确度明显降低。遗传距离较近的亚群间(第I亚群和第II亚群)预测准确度优于遗传距离远的亚群间的预测准确度(第I亚群与第III亚群和第II亚群与第III亚群)。但是也有例外, 用第II亚群预测第III亚群时, 预测准确度为0.21, 超过其他亚群间百粒重预测准确度。
以任意2个亚群为训练群体时, 估算剩余亚群百粒重预测准确度发现, 以第I、第III亚群为训练群体, 第II亚群为验证群体时预测准确度最高为0.29; 当以第II, III亚群为训练群体, 第I亚群为验证群体时, 百粒重的预测准确度最低为0.07。比较结果发现, 当第II亚群与第III亚群分别处于训练群体和验证群体, 能提高百粒重的预测准确度。
3 讨论
大豆育成品种的群体结构是在长期的人工选择过程中形成的, 反映了不同时期、不同生态类型大豆品种间的育种目标的差异。明确大豆品种群体结构对基于关联分析方法的基因挖掘具有十分重要的意义。前人研究表明, 我国大豆育成品种及野生资源群体均存在复杂的遗传结构, 并且与大豆品种(种质)的地理来源密切相关[34-36]。宋喜娥等利用248份栽培大豆微核心种质也获得了相似的结果[37]。张军等[38]研究表明, 中国大豆育成品种群体在遗传结构上具有地理生态分化和育成时期分化, 不同亚群具有相对遗传特异性。基于贝叶斯理论的Structure软件和主成分分析方法成为群体结构分析的主要手段[39-40]。
本研究利用Structure软件明确了参试品种的群体结构, 并得到主成分分析方法验证。供试群体被划分为3个亚群, 在第一主成分上, 不同生态类型品种被分成两部分, 在第二主成分上, 不同省份品种被分开。这与前人研究中指出的大豆品种群体遗传结构与地理生态分类相关的结论[34-38]相同。引进的日本品种十胜长叶是我国大豆育成品种中重要的核心亲本[41]。在本研究中, 十胜长叶被划分到北方春大豆的第一亚群中, 这与郭娟娟等[41]的研究结果相似。
利用模拟数据和试验数据研究表明, 训练群体的构成以及训练群体与候选群体的关系是影响全基因组选择预测准确度的重要因素[14, 20, 42-44]。训练群体与候选群体遗传关系越紧密, 预测准确度越高[14, 43]; 训练群体和候选群体存在相同的群体结构能提高全基因组选择的预测准确度[45]; 目标性状的遗传结构也能够通过群体结构对全基因组预测准确度造成影响[14]。本研究比较了供试总体以及不同亚群内的百粒重的预测准确度, 发现在目标性状变异系数大的群体中, 全基因组选择的预测准确度高。百粒重在供试总体和第I、第II、第III亚群中的变异系数分别为7.80%、6.53%、7.56%和10.49%, 相应百粒重的预测准确度分别为0.54、0.24、0.51和0.75。说明训练群体中丰富的遗传变异能够提高大豆百粒重的预测准确度。这与前人研究结果一致[42-43, 45]。
Habier等[46]将基因组选择的预测准确度来源归结为标记与QTL的连锁不平衡和训练群体与候选群体的遗传关系2个部分, 当训练群体和验证群体遗传距离较远时, 目标性状的预测准确度主要依靠标记与QTL的连锁不平衡关系。在本研究中第II亚群与第III亚群遗传距离最远, 但是以第II亚群预测第III亚群的百粒重的预测准确度在不同亚群间最高。这可能是由于在第II亚群和第III亚群间标记与百粒重的QTL具有相似的连锁不平衡状态。Asoro等[43]研究表明, 目标性状、群体构成方式和全基因组选择模型及其互作均能对预测准确度产生影响。本研究只用随机回归最佳线性无偏预测方法估算了群体构成方式对大豆百粒重全基因组选择预测准确度的影响, 在进一步研究中可以考虑比较不同选择模型对大豆百粒重预测准确度的作用。Guo等[44]利用来自于28个国家的413份水稻品种研究表明, 当训练群体与验证群体存在相同的群体结构时有利于全基因组预测准确度的提高。在本研究中, 基于群体结构取样产生的群体预测准确度相比于随机取样的预测准确度提高了2.34%, 差异不显著。
本研究所用的标记数量远高于群体数量, 符合全基因组选择理论[8-11]。同时基于已发表的大豆重要农艺性状的QTL的位置选择标记, 保证了对大豆基因组的覆盖程度。利用随机回归最佳线性无偏预测模型获得的大豆百粒重的预测准确度平均值为0.539。Shu等[16]利用79个SCAR标记和288份大豆比较随机回归最佳线性无偏预测模型和贝叶斯线性回归模型对大豆百粒重的预测准确度发现, 在不同预测模型间的预测准确度平均值分别为0.692和0.690, 高于本研究获得的预测准确度。Shu等[16]所用的标记数量较少, 但预测准确度比本研究结果高, 可能与标记中包括与大豆百粒重显著相关6个的SCAR标记有关。Bao等[17]也发现, 在标记密度较低的情况下(96个), 将与目标性状相关的标记作为固定效应, 其他标记作为随机效应时, 可将大豆抗胞囊线虫病的全基因组选择预测准确度提高20%。因此, 今后可以通过关联分析筛选与百粒重相关的标记并建立适宜的预测模型, 提高大豆百粒重的全基因组选择预测准确度。
4 结论
利用随机取样方法和基于群体结构的取样方法构建训练群体, 以随机回归最佳线性无偏预测模型和5倍交互验证的方法对大豆百粒重进行全基因组选择, 预测准确度分别为0.5387和0.5513。群体构成方式与百粒重全基因组选择预测准确度关系密切, 以亚群内显著高于亚群间、遗传距离近群体优于遗传距离远群体; 提高训练群体百粒重的遗传变异程度能显著提高预测准确度。本研究结果为大豆重要性状选择提供了一种分子育种新方法。
附表 请见网络版: 1) 本刊网站http://zwxb.chinacrops. org/; 2) 中国知网http://www.cnki.net/; 3) 万方数据http://c.wanfangdata.com.cn/Periodical- zuowxb.aspx。
[1] 盖钧镒, 熊冬金, 赵团结. 中国大豆育成品种系谱与种质基础(1923–2005). 北京: 中国农业出版社, 2015. pp 11–12 Gai J Y, Xiong D J, Zhao T J. The Pedigrees and Germplasm Bases of Soybean Cultivars Released in China (1923–2005). Beijing: China Agriculture Press, 2015. pp 11–12 (in Chinese)
[2] 徐东河, 李东艳, 程舜华. 大豆百粒重与抗旱性及产量的关系. 中国油料, 1991, (3): 64–66 Xu D H, Li D Y, Cheng S H. Relationship between 100-seed weight and anti-draught and yield of soybean., 1991, (3): 64–66 (in Chinese)
[3] 王占廷, 栾素荣, 程舜华. 大豆百粒重与产量的相关分析. 大豆通报, 1997, (2): 9 Wang Z T, Luan S R, Cheng S H. Relationship analysis between 100-seed weight and yield in soybean., 1997, (2): 9 (in Chinese)
[4] 汪霞, 徐宇, 李广军, 李河南, 艮文全, 章元明. 大豆百粒重QTL定位. 作物学报, 2010, 36: 1674–1682 Wang X, Xu Y, Li G J, Li H N, Gen W Q, Zhang Y M. Mapping quantitative trait loci for 100-seed weight in soybean (L. Merr.)., 2010, 36: 1674–1682 (in Chinese with English abstract)
[5] 陈庆山, 蒋洪蔚, 孙殿君, 刘春燕, 辛大伟, 曾庆力, 马占洲, 胡国华. 利用野生大豆染色体片段代换系定位百粒重QTL. 大豆科学, 2014, 33: 154–160 Chen Q S, Jiang H W, Sun D J, Liu C Y, Xin D W, Zeng Q L, Ma Z Z, Hu G H. QTL Mapping for 100-seed weight using wild soybean chromosome segment substitution lines., 2014, 33: 154–160 (in Chinese with English abstract)
[6] 张英虎, 孟珊, 贺剑波, 王宇峰, 邢光南, 赵团结, 盖钧镒. 大豆重组自交系群体NJRSXG百粒重超亲分离的遗传解析. 中国农业科学, 2015, 48: 4408–4416 Zhang Y H, Meng S, He J B, Wang Y F, Xing G N, Zhao T J, Gai J Y. The genetic constitution of transgressive segregation of the 100-seed weight in a recombinant inbred line population NJRSXG of soybean., 2015, 48: 4408–4416 (in Chinese with English abstract)
[7] 齐照明, 孙亚男, 陈立君, 郭强, 刘春燕, 胡国华, 陈庆山. 基于Meta分析的大豆百粒重的QTLs定位. 中国农业科学, 2009, 42: 3795–3803 Qi Z M, Sun Y N, Chen L J, Guo Q, Liu C Y, Hu G H, Chen Q S. Meta-analysis of 100-seed weight QTL in soybean., 2009, 42: 3795–3803 (in Chinese with English abstract)
[8] Goddard M E, Hayes B J. Genomic selection., 2007, 124: 323–330
[9] Jannink J L, Lorenz A J, Iwata H. Genomic selection in plant breeding: from theory to practice., 2010, 9: 166–177
[10] Nakaya A, Isobe S N. Will genomic selection be a practical method for plant breeding?, 2012, 110: 1303–1316
[11] Meuwissen T H E, Hayes B J, Goddar M E. Prediction of total genetic value using genome-wide dense marker maps., 2001, 157: 1819–1829
[12] Zhao Y, Gowda M, Liu W, Wurschum T, Maurer H P, Longin F H, Ranc N, Reif J C. Accuracy of genomic selection in European maize elite breeding populations., 2012, 124: 769–776
[13] Zhao Y, Gowda M, Longin F H, Wurschum T, Ranc N, Reif J C. Impact of selective genotyping in the training population on accuracy and bias of genomic selection., 2012, 125: 707–713
[14] Crossa J, Perez P, Hickey J, Burgueno J, Ornella L, Rojas J C, Zhang X, Dreisigacker S, Babu R, Li Y, Mathews K. Genomic prediction in CIMMYT maize and wheat breeding programs., 2014, 112: 48–60
[15] Dawson J C, Endelman J B, Heslot N, Crossa J, Poland J, Dreisigacker S, Manes Y, Sorrells M E, Jannink J L. The use of unbalanced historical data for genomic selection in an international wheat breeding program., 2013, 154: 12–22
[16] Shu Y J, Yu D S, Wang D, Bai X, Zhu Y M, Guo C H. Genomic selection of seed weight based on low-density SCAR markers in soybean., 2013, 12: 2178–2188
[17] Bao Y, Vuong T, Meinhardt C, Tiffin P, Denny R, Chen S Y, Nguyen H T, Orf J H, Young N D. Potential of association mapping and genomic selection to explore PI88788 derived soybean cyst nematode resistance., 2014, 7: 1–13
[18] Sprdel J, Begum H, Akdemir D, Virk P, Collard B, Redona E, Atlin G, Jannink J L, McCouch S R. Genomic selection and association mapping in rice (): effect of trait genetic architecture, training population composition, marker number and statistical model on accuracy of rice genomic selection in elite, tropical rice breeding lines., 2015, 11: e1004982
[19] Zhong S Q, Dekkers J C, Fernando R L, Jannink J L. Factors affecting accuracy from genomic selection in population derived from multiple inbred lines: a barley case study., 2009, 182: 355–364
[20] Wang Y, Mette M F, Miedaner T, Gottwald M, Wilde P, Rif J C, Zhao Y S. The accuracy of prediction of genomic selection in elite hybrid rye populations surpasses the accuracy of marker- assisted selection and is equally augmented by multiple field evaluation locations and test years., 2014, 15: 556–567
[21] Reif J C, Zhao Y S, Wurschum T, Gowda M, Hahn V. Genomic selection of sunflower hybrid performance., 2013, 132: 107–114
[22] Denis M, Bouvet J M. Efficiency of genomic selection with models including dominance effect in the context ofbreeding., 2013, 9: 37–51
[23] Desta Z A, Ortiz R. Genomic selection: genome-wide prediction in plant improvement., 2014, 19: 592–601
[24] Heslot N, Jannink J L, Sorrells M E. Perspective for genomic selection applications and research in plants., 2015, 55: 1–12
[25] Schmutz J, Cannon S B, Schlueter J, Ma J X, Mitros T, Nelson W, Hyten D L, Song Q J, Thelen J J, Cheng J L, Xu D, Hellsten U, May G D, Yu Y S, Sakurai T, Umezawa T S, Bhattacharyya M K, Sandhu D, Valliyodan B, Lindquist E, Peto M, Grant D, Shu S Q, Goodstein D, Barry K, Griggs M F, Abernathy B, Du J C, Tian Z X, Zhu L C, Gill N, Joshi T, Libault M, Sethuraman A, Zhang X C, Shinozaki K, Nguyen H T, Wing R A, Cregan P, Specht J, Grimwood J, Rokhsar D, Stacey G, Shoemaker R C, Jachson S A. Genome sequence of the palaeoployploid soybean., 2010, 463: 178–183
[26] Lam H M, Xu X, Liu X, Chen W B, Yang G H, Wong F L, Li M W, He W M, Qin N, Wang B, Li J, Jian M, Wang J, Shao G H, Wang J, Sun S S, Zhang G Y. Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection., 2010, 42: 1053–1059
[27] Li Y H, Zhou G Y, Ma J X, Jiang W K, Jin L G, Zhang Z H, Guo Y, Zhong J B, Sui Y, Zheng L T, Zhang S S, Zou Q Y, Shi X H, Li Y F, Zhang W K, Hu Y Y, Kong G Y, Hong H L, Tan B, Song J, Liu Z X, Wang Y S, Ruan H, Yeung C K, Liu J, Wang H L, Zhang L J, Guan R X, Wang K J, Li W B, Chen S Y, Chang R Z, Jiang Z, Jackson S A, Li R Q, Qiu L J. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits., 2014, 32: 1045–1052
[28] Zhou Z K, Jiang Y, Wang Z, Gou Z H, Lyu J, Li W Y, Yu Y J, Shu L Q, Zhao Y J, Ma Y M, Fang C, Shen Y T, Liu T F, Li C C, Li Q, Wu M, Wang M, Wu Y S, Dong Y, Wan W T, Wang X, Ding Z L, Gao Y D, Xiang H, Zhu B G, Lee S H, Wang W, Tian Z X. Re-sequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean., 2015, 33: 408–414
[29] Song Q J, Hyten D L, Jia G F, Quigley C V, Fickus E W, Nelson R L, Cregan P B. Development and evaluation of SoySNP50K, a high-density genotyping array for soybean., 2013, 8: e54985
[30] 邱丽娟, 常汝镇, 刘章雄, 关荣霞, 李英慧. 大豆种质资源描述规范和数据标准. 北京: 中国农业出版社, 2006. pp 18–24 Qiu L J, Chang R Z, Liu Z X, Guan R X, Li Y H. Descriptors and Data Standard for Soybean (spp.). Beijing: China Agriculture Press, 2015. pp 18–24 (in Chinese)
[31] Fehr W R. Genetic contributions to yield gains of five major crop plants; proceedings of a symposium sponsored by Division C-1 of the Crop Science Society of America, in Atlanta, Georgia- ResearchGate, 1984.
[32] Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study., 2005, 14: 2611–2620
[33] Bradbury P J, Zhang Z W, Kroon D E, Casstevens T M, Ramdoss Y, Buckler E S. TASSEL: software for association mapping of complex traits in diverse samples., 2007, 23: 2633–2635
[34] 文自翔, 赵团结, 郑永战, 刘顺湖, 王春娥, 王芳, 盖钧镒. 中国栽培和野生大豆农艺品质性状与SSR标记的关联分析: I. 群体结构及关联标记. 作物学报, 2008, 34: 1169–1178 Wen Z X, Zhao T J, Zheng Y Z, Liu S H, Wang C E, Wang F, Gai J Y. Association analysis of agronomic and quality traits with SSR markers inandin China: I. Population structure and associated markers., 2008, 34: 1169–1178 (in Chinese with English abstract)
[35] 张军, 赵团结, 盖钧镒. 中国东北大豆育成品种遗传多样性和群体遗传结构分析. 作物学报, 2008, 34: 1529–1536 Zhang J, Zhao T J, Gai J Y. Genetic diversity and genetic structure of soybean cultivar population released in Northeast China., 2008, 34: 1529–1536 (in Chinese with English abstract)
[36] 范虎, 赵团结, 丁艳来, 邢光南, 盖钧镒. 中国野生大豆群体特征和地理分化的遗传分析. 中国农业科学, 2012, 45: 414–425 Fan H, Zhao T J, Ding Y L, Xing G N, Gai J Y. Genetic analysis of the characteristics and geographic differentiation of Chinese wild soybean population., 2012, 45: 414–425 (in Chinese with English abstract)
[37] 宋喜娥, 李英慧, 常汝镇, 郭平毅, 邱丽娟. 中国栽培大豆((L.) Merr.) 微核心种质的群体结构与遗传多样性. 中国农业科学, 2010, 43: 2209–2219 Song X E, Li Y H, Chang R Z, Guo P Y, Qiu L J. Population sturcture and genetic diversity of mini core collection of cultivated soybean ((L.) Merr.) in China., 2010, 43: 2209–2219 (in Chinese with English abstract)
[38] 张军, 赵团结, 盖钧镒. 中国大豆育成品种群体遗传结构分化和亚群特异性分析. 中国农业科学, 2009, 42: 1901–1910 Zhang J, Zhao T J, Gai J Y. Analysis of genetic structure differentiation of released soybean cultivar population and specificity of subpopulations in China., 2009, 42: 1901–1910 (in Chinese with English abstract)
[39] 魏世平, 刘晓芬, 杨胜先, 吕海燕, 牛远, 章元明. 中国栽培大豆群体结构不同分类方法的比较. 南京农业大学学报, 2011, 34(2): 13–17 Wei S P, Liu X F, Yang S X, Lyu H Y, Niu Y, Zhang Y M. Comparison of various clustering methods for population structure in Chinese cultivated soybean ((L.) Merr.)., 2011, 34(2): 13–17 (in Chinese with English abstract)
[40] 黎裕, 李英慧, 杨庆文, 张锦鹏, 张金梅, 邱丽娟, 王天宇. 基于基因组学的作物种质资源研究: 现状与展望. 中国农业科学, 2015, 48: 3333–3353 Li Y, Li Y H, Yang Q W, Zhang J P, Zhang J M, Qiu L J, Wang T Y. Genomics-based crop germplasm research: advances and perspectives., 2015, 48: 3333–3353 (in Chinese with English abstract)
[41] 郭娟娟, 常汝镇, 章建新, 张巨松, 关荣霞, 邱丽娟. 日本大豆种质十胜长叶对我国大豆育成品种的遗传贡献分析. 大豆科学, 2007, 26: 807–819 Guo J J, Chang R Z, Zhang J X, Zhang J S, Guan R X, Qiu L J. Contribution of Japanese soybean germplasm TOKACHI- NAGAHA to Chinese soybean cultivars., 2007, 26: 807–819 (in Chinese with English abstract)
[42] Toosi A, Fernando R L, Dekkers J C M. Genomic selection in admixed and crossbred populations., 2010, 88: 32–46
[43] Asoro F G, Newell M A, Beavis W D, Scott M P, Jannink J L. Accuracy and training population design for genomic selection on quantitative traits in elite North American oats., 2011, 4: 132–144
[44] Guo Z G, Tucker D M, Basten C J, Gandhi H, Ersoz E, Guo B H, Xu Z Y, Wang D L, Gay G. The impact of population structure on genomic prediction in stratified populations., 2014, 127: 749–762
[45] Daetwyler H D, Wong R P, Villanueva B, Woolliams J A. The impact of genetic architecture on genome-wide evaluation methods., 2010, 185: 1021–103
[46] Habier D, Fernando R L, Dekkers J C M. Impact of genetic relationship information on genome-assisted breeding values., 2007, 177: 2389–2397
Effect of Population Structure on Prediction Accuracy of Soybean 100-Seed Weight by Genomic Selection
MA Yan-Song1,2,13, LIU Zhang-Xiong1, WEN Zi-Xiang3, WEI Shu-Hong4, YANG Chun-Ming5, WANG Hui-Cai6, YANG Chun-Yan7, LU Wei-Guo8, XU Ran9, ZHANG Wan-Hai10, WU Ji-An11, HU Guo-Hua12, LUAN Xiao-Yan13, FU Ya-Shu14, GUO Tai15, WANG Shu-Ming5, HAN Tian-Fu1, ZHANG Meng-Chen7, ZHANG Lei16, YUAN Bao-Jun17, GUO Yong1, Jochen C. REIF18, JIANG Yong18, LI Wen-Bin2, WANG De-Chun3, and QIU Li-Juan1,*
1National Key Facility for Crop Gene Resources and Genetic Improvement / Institute of Crop Sciences, Chinese Academy of Agricultural Sciences, Beijing 100081, China;2College of Agriculture, Northeast Agricultural University, Harbin 150030, Heilongjiang, China;3Department of Plant, Soil and Microbial Sciences, Michigan State University, East Lansing MI 48824, USA;4Institute of Crop Breeding, Heilongjiang Academy of Agricultural Sciences, Harbin 150086, Heilongjiang, China;5Soybean Research Institute, Jilin Academy of Agricultural Sciences, Changchun 130033, Jilin, China;6Chifeng Institute of Agricultural Sciences, Chifeng 024031, Inner Mongolia, China;7Institution of Cereal and Oil Crops, Hebei Academy of Agriculture and Forestry Sciences, Shijiazhuang 050031, Hebei, China;8Economic Crops Institute, Henan Academy of Agricultural Sciences, Zhengzhou 450002, Henan, China;9Crop Research Institute, Shandong Academy of Agricultural Sciences, Jinan 250010, Shandong, China;10Hulunbeier Institute of Agricultural Sciences, Hulunbeier 021000, Inner Mongolia, China;11Heihe Branch Institute, Heilongjiang Academy of Agricultural Sciences, Heihe 164300, Heilongjiang, China;12Crop Research and Breeding Center of Land-Reclamation, Harbin 150090, Heilongjiang, China;13Soybean Research Institute, Heilongjiang Academy of Agricultural Sciences, Harbin 150086, Heilongjiang, China;14Suihua Branch Institute, Heilongjiang Academy of Agricultural Sciences, Suihua 152052, Heilongjiang, China;15Jiamusi Branch Institute, Heilongjiang Academy of Agricultural Sciences, Jiamusi 154007, Heilongjiang, China;16Crop Research Institute, Anhui Academy of Agricultural Sciences, Hefei 230031, Anhui, China;17Zhoukou Institute of Agricultural Sciences, Zhoukou 466001, Henan, China;18Department of Breeding Research, Leibniz Institute of Plant Genetics and Crop Plant Research (IPK), Gatersleben 06466, Germany
Hundred-seed weight is an important yield component and has positive relationship with soybean yield under certain conditions. The genetic gain of 100-seed weight based on traditional breeding or markers assisted-selection is limited because it is controlled by plenty of small effect genes. Genomic selection offers an approach to accelerate the soybean 100-seed weight breeding. However, the effect of population structure on soybean 100-seed weight prediction accuracy has not been elaborated. In our study 280 soybean varieties with phenotypic data evaluated in multi-location in 2008–2012 and 5361 SNPs genotype were used to explore the effect of population structure on 100-seed weight prediction accuracy. The best linear unbiased prediction of 100-seed weight of each variety was calculated according to mixed linear model. Ridge regression best linear unbiased prediction and five-fold cross validation were used to estimate the 100-seed weight prediction accuracy. Our research showed that the range of 100-seed weight, which was from –0.15 to +0.75. Hundred-seed weight prediction accuracy was affected by population structure significantly. The prediction accuracy within a subset (+0.24 to +0.75) was higher than that between subsets (-0.15 to +0.29). When the genetic distance between subsets increased from 0.1566 to 0.2201, the 100-seed weight prediction accuracy was decreased by 27.87%. Compared with random sampling training population, the training population composed based on genetic structure improved 100-seed weight prediction accuracy by 2.34%. In summary, we are clear about the soybean 100-seed weight genomic selection accuracy and the effect of population structure on genomic selection accuracy. The genomic selection is an efficient method to improve the soybean breeding.
; 100-seed weight; genomic selection; prediction accuracy; genetic structure
2017-02-10;
2017-09-10;
2017-10-30.
10.3724/SP.J.1006.2018.00043
通信作者(Corresponding author):邱丽娟, E-mail: qiulijuan@caas.cn, Tel: 010-82105840
E-mail: mys771007@hotmail.com
本研究由国家转基因生物新品种培育重大专项(2014ZX08004001)和中国农业科学院农业科技创新项目资助。
This study was supported by the National Major Project for Developing New GM Crops (2014ZX08004001) and the Agricultural Science and Technology Innovation Program (ASTIP) of Chinese Academy of Agricultural Sciences.
URL: http://kns.cnki.net/kcms/detail/11.1809.S.20171030.0858.002.html
猜你喜欢
现代临床医学(2023年1期)2023-03-24 08:30:06
中国医药科学(2022年5期)2022-05-05 23:58:07
今日农业(2021年11期)2021-08-13 08:53:24
建筑科技(2018年6期)2018-08-30 03:40:54
中国交通信息化(2016年5期)2016-06-06 03:51:43
中国卫生标准管理(2015年3期)2016-01-14 03:41:45
安徽医药(2014年4期)2014-03-20 13:13:40
遗传(2014年3期)2014-02-28 20:58:49
天津冶金(2014年4期)2014-02-28 16:52:58
世界科学(2014年8期)2014-02-28 14:58:31
