
控制普通野生稻种子休眠性QTL的定位
2018-01-18 01:29:15孙爱伶伍洪铭陈高明张天雨曹鹏辉刘世家万建民
作物学报 2018年1期
孙爱伶 伍洪铭 陈高明 张天雨 曹鹏辉 刘世家 江 玲,* 万建民,2
控制普通野生稻种子休眠性QTL的定位
孙爱伶1伍洪铭1陈高明1张天雨1曹鹏辉1刘世家1江 玲1,*万建民1,2
1南京农业大学作物遗传与种质创新国家重点实验室 / 农业部长江中下游粳稻生物学与遗传育种重点实验室 / 江苏省植物基因工程技术研究中心, 江苏南京 210095;2中国农业科学院作物科学研究所 / 作物基因资源与遗传改良国家重大科学工程, 北京 100081
水稻种子休眠性是关系到稻米品质和稻种质量的一个重要农艺性状。研究水稻种子休眠性遗传及分子机制对培育具有适度休眠性的优良水稻品种具有重要意义。本研究以籼稻品种9311为受体、普通野生稻为供体的染色体片段置换系群体为材料, 在后熟不同时间检测群体种子休眠性, 对控制种子休眠性的QTL进行定位分析, 共定位到14个QTL, 分布在第3、第4、第5、第6、第7、第10、第11、第12染色体上。筛选休眠性显著强于背景亲本9311的家系, 分析这些家系携带的QTL数目, 表明携带的位点越多, 休眠性越强。进一步利用家系Q14与9311的F2群体验证了第7染色体标记RM180和RM21323之间存在一个效应较大的QTL, 该位点LOD值为18.49, 可解释的表型变异率为33.53%, 表明该位点是一个控制普通野生稻种子休眠性的主效QTL, 且能稳定遗传。本研究为野生稻种子休眠基因的精细定位及克隆奠定了基础, 且为培育强休眠性籼稻品种提供了育种材料。
野生稻; 种子休眠; 染色体片段置换系; QTL
种子休眠是指完好无损有活力的种子在适宜萌发的条件下(光照、温度、水分、氧气等)不能发芽的现象[1]。水稻种子休眠性是一个重要的农艺性状。一方面休眠性较弱的品种在成熟后期遭遇高温多雨天气, 容易发生穗发芽, 从而降低稻米的品质和种子质量[2]; 另一方面休眠性过强的品种, 可能导致播种时田间出苗率低, 出苗不整齐, 不利于水稻直播的推广应用。为培育适度休眠性的优良品种, 研究水稻种子休眠性的遗传特性和分子机制, 显得尤为重要。
水稻种子休眠性是一个十分复杂的数量性状, 影响种子休眠的因素包括内在因素和外在环境因素,其中内在因素主要含植物激素、胚效应、种皮效应等; 外在因素主要有温度、水分、空气、光照等。随着分子标记技术的发展, 水稻高密度遗传图谱的构建使得对这些数量性状位点的定位及克隆成为可能。迄今, 已初步定位了100多个与水稻休眠性有关的QTL, 广泛分布于水稻12条染色体上, 其中在第1、第3、第5、第6、第7和第11染色体上的休眠QTL较多[3], 且检测到一些稳定表达的种子休眠性QTL, 并对其进行了精细定位和克隆。例如, 日本学者Yano课题组利用日本晴和Kasalath衍生的高代回交群体(BC4F2), 将位于第7染色体的QTL进行了图位克隆, 这是首个图位克隆的控制水稻种子休眠性的基因, 但其调控休眠的分子机制尚不清楚[4]。Takeuchi等[5]利用高代回交群体对位于第3染色体控制种子休眠性的QTL进行了精细定位, 将其限制在RFLP标记R10942与C2045之间的区域内, 与标记C1488共分离。Gu等检测到了一个来自杂草稻的位于第12染色体的控制种子休眠的主效QTL, 进一步精细定位将其限定在标记RM28642与SD12m50之间的小于75 kb的区间内[6-7]。Lu等[8]利用南粳35和N22衍生的高代回交群体, 定位到2个控制N22种子休眠的QTL和, 将位于第1染色体的限定在标记RM11669和RM1216之间, 将位于第5染色体的限定在标记RM480和RM3664之间。罗正良[9]利用近等基因系K7481 ()与背景亲本冈46B的F2群体, 将位于第8染色体控制种子穗发芽抗性的QTL精细定位在标记RM23511和RM23520的103 kb之间。
野生稻由于长期处于野生状态, 经受了各种灾害和不良环境的自然选择, 具有抗病、耐逆、休眠性强等多种特性, 是天然的基因库, 保持有栽培稻不具有或已经消失了的基因, 遗传多样性丰富, 是栽培稻育种的宝贵种质资源, 也是研究稻种起源、演变和分化必不可少的材料[10]。为此, 中国农业科学院作物科学研究所杨庆文课题组利用普通野生稻与9311构建了以9311为受体, 野生稻为供体的一套染色体片段置换系群体, 该群体为挖掘野生稻中控制种子休眠性的基因提供了基础。
1 材料与方法
1.1 供试材料
以9311为受体、普通野生稻为供体的染色体片段置换系(CSSL)群体, 共200个家系由杨庆文课题组提供。
2015年正季在南京农业大学土桥水稻试验基地种植CSSL群体及亲本9311 (普通野生稻在南京不能抽穗)。每个家系2行, 每行10株。行距为20.0 cm, 株距为13.3 cm, 每穴1株。田间的水肥管理同常规大田。抽穗后35 d收获各家系种子, 选择每个家系3株长势一致的植株分单株收种, 在室温条件下后熟21 d和28 d后检测种子的休眠性。同时, 从CSSL群体各家系中挑选典型单株与9311杂交。2015年冬季在海南加代, 自交得到F2种子。
根据正季种子休眠性鉴定的结果, 选择强休眠性的9个家系以及强休眠性Q14家系与9311杂交自交的F2群体(含275个单株)和亲本9311, 于2016年正季种植在土桥水稻试验基地, 种植方法同2015年。抽穗后35 d收获F2群体各单株种子, 在室温条件下后熟14 d后检测种子的休眠性。同样在抽穗后35 d收获9个强休眠性的CSSL家系, 立即做发芽试验检测种子休眠性, 并在室温条件下后熟14 d后再次检测种子的休眠性。
1.2 种子休眠性的表型鉴定
参照Wan等[11]的方法, 略作修改, 鉴定种子休眠性表型。从CSSL群体各单株随机数取50粒健康饱满的种子, 放入铺有两层滤纸的9 cm培养皿, 加水置30°C培养箱黑暗条件下发芽。以胚根或胚芽超过半粒种子长为发芽标准, 统计第7天的种子发芽率。以3个单株的发芽率平均数作为家系的发芽率表型值, 进而判断各家系种子的休眠性。
从F2群体各单株随机数取50粒健康饱满的种子置双层湿润滤纸上, 设2个重复, 在30°C黑暗条件下发芽, 按上述标准检测第7 d发芽率, 以种子发芽率来评价种子休眠性。
1.3 DNA的提取及分子标记分析
在Q14/9311 F2群体分蘖期取各单株叶片1张。采用CTAB法[12], 稍作修改, 提取DNA。将提取的DNA加200 µL ddH2O溶解作为DNA母液, 用Perkin Elmer MBA 2000 DNA浓度测定仪测定母液浓度, 并稀释成20 ng µL–1的DNA工作液, 作为PCR扩增反应的模板, 4°C冰箱中保存。
PCR体系包括DNA模板1 µL (20 ng µL–1), 引物1.2 µL (2 mmol L–1), dNTP 0.4 µL (2.5 mmol L–1), buffer (Mg2+) 1 µL, ddH2O 6.6 µL, rDNA聚合酶0.1 µL (0.1 U)。PCR扩增程序为95°C预变性5 min, 94°C变性30 s, 55°C退火30 s, 72°C延伸40 s, 变性–退火–延伸步骤进行35次循环, 72°C10 min, 4°C保存。扩增产物经8%非变性聚丙烯酰胺凝胶电泳分离, 参考Sangnietti等[13]的方法银染显色。
1.4 遗传图谱构建及休眠性QTL检测
CSSL群体的分子遗传图谱(基因型背景)由本实验室陈高明博士提供, 是用3410个覆盖全基因组的SNP标记对群体进行基因型分析(SNP标记数据等信息另文发表)。利用QTL IciMapping 4.1软件[14], 采用逐步回归相似比率测试(RSTEP-LRT)方法, 进行种子休眠性QTL检测。
Q14和9311衍生的F2群体的图谱构建, 用覆盖全基因组的223个SSR和Indel标记来确定家系Q14的置换片段位置, 部分置换主要在第7染色体。利用第7染色体亲本间多态性较好的4个SSR标记和4个Indel标记, 分析F2分离群体中275个单株的基因型, 获得每个单株的分子数据。利用QTL IciMapping 4.1软件[14], 采用复合区间作图方法对F2群体进行种子休眠性QTL检测。以LOD值2.5作为阈值判断QTL是否存在。遵循McCouch等[15]的方法命名QTL。用于分析Q14/9311 F2群体各单株基因型的标记序列信息见表1。
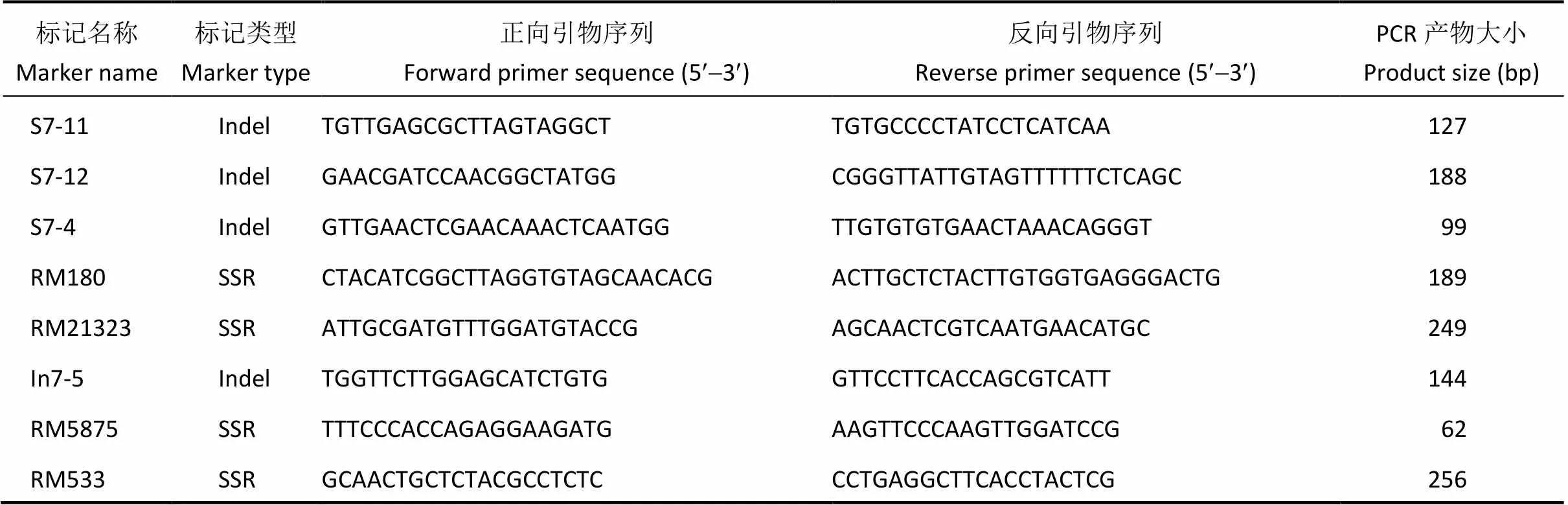
表1 用于分析Q14/9311 F2群体各单株基因型的SSR和Indel引物序列
SSR: 简单重复序列; Indel: 插入缺失。SSR: simple sequence repeat; Indel: insertion/deletion.
2 结果与分析
2.1 CSSL群体休眠性表型分析
2.1.1 2015年正季群体种子后熟21 d 普通野生稻有较强的种子休眠性, 而9311的种子休眠性相对较弱。对后熟过程中亲本9311的种子, 每隔3 d做一次发芽试验, 检测其休眠性。当9311的发芽率达到中等水平, 即后熟21 d时, 其发芽率为(75±2)%, CSSL群体的发芽率呈0~100%的连续分布, 且表现出超亲现象(图1)。有20%家系的发芽率在70%以下,显著低于背景亲本9311, 表明这些休眠性很强的家系中携带有普通野生稻增强休眠性的基因位点。
2.1.2 2015年正季群体种子后熟28 d 背景亲本9311的发芽率达到(94±3)%, 而CSSL群体的发芽率仍呈连续分布, 变异幅度在10%~100%之间(图2)。与后熟21 d相比, 群体内60%的家系的发芽率已达到90%以上, 但仍有30%家系的发芽率显著低于背景亲本9311。表明这些家系存在更强的休眠性基因位点。

图1 2015年CSSL群体种子后熟21 d后休眠性次数分布图
Germination rate (%)表示种子在第7天的发芽率, 每个家系3个重复, 每个重复50粒; No. of lines为对应发芽率的家系数, 用柱子上的数字表示。
Germination rate (%) indicates that at seventh days of germination, three plants for each line were tested, using 50 grains for each replicate; No. of lines in the line number at the corresponding germination rate, showing on the column.
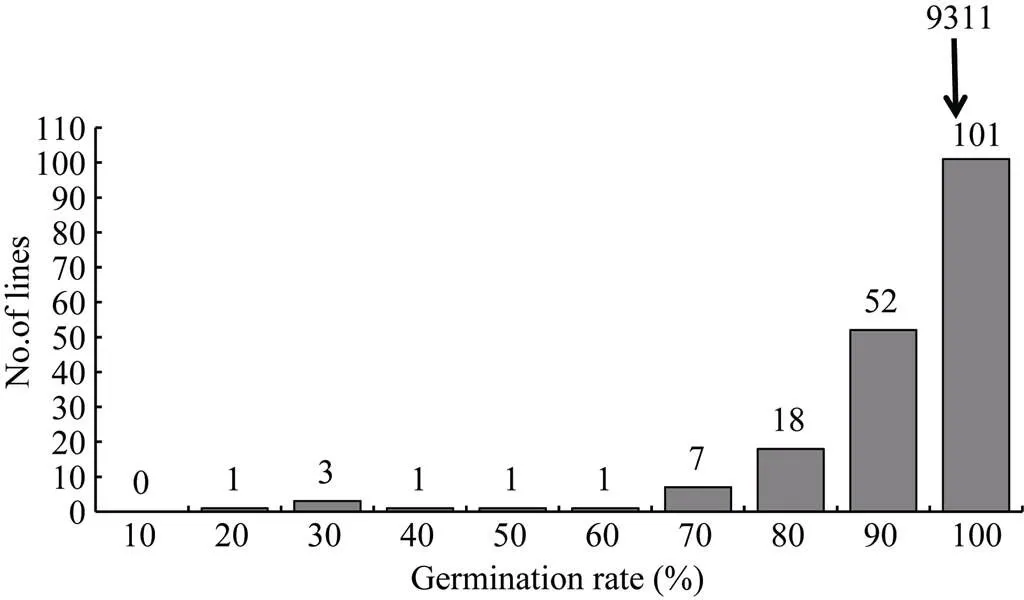
图2 2015年CSSL群体种子后熟28 d后休眠性次数分布图
Germination rate (%)表示种子在第7天的发芽率, 每个家系3个重复, 每个重复50粒; No. of lines为对应发芽率的家系数, 用柱子上的数字表示。
Germination rate (%) indicates that at seventh days of germination, three plants for each line were tested, using 50 grains for each replicate; No. of lines in the line number at the corresponding germination rate, showing on the column.
2.2 CSSL群体种子休眠性QTL检测
2.2.1 2015年正季群体种子后熟21 d 在CSSL群体检测到6个休眠性QTL, 分别位于第4、第7、第11、第12染色体上, 其中于第7、第12染色体均检测到2个位点。LOD值介于2.75~13.01之间, 贡献率在3.93~22.38之间, 其中的LOD值最大, 贡献率也最高, 达到22.38%, 其加性效应为-21.62, 表明该位点上来自野生型的等位基因能降低21%的发芽率(表2)。
2.2.2 2015年正季群体种子后熟28 d 在CSSL群体共检测到11个QTL, 分别位于第3、第5、第6、第7、第10、第11、第12染色体上, LOD值介于3.07~20.87之间, 贡献率在1.74~14.75之间。其中在第3染色体上检测到2个位点, 而第12染色体上检测到4个位点, 其中和在后熟21 d的条件下也被检测到, 说明这2个位点的真实性。而另外2个位点和是被新检测到的, 其真实性有待进一步验证。和两个位点距离很近且控制种子休眠性的基因来源不同, 如果这2个位点是真实存在的, 说明在普通野生稻的该区间存在作用相反的2个位点。的LOD值最大, 贡献率也最高, 达到20.87%, 加性效应为–14.20, 表明该位点来自野生稻的等位基因能降低14%的发芽率(表3)。
在不同后熟时间检测到3个相同的位点,、和。此外,和位置很近, 猜测可能是同一个休眠性QTL。表明这4个位点控制的休眠性在后熟过程中不易被打破。另外, 在后熟28 d时检测到更多的位点, 其可能原因是, 背景亲本9311有一定休眠性, 且其休眠性更易被解除, 后熟28 d时, 其发芽率达到94%, 而CSSL群体中比9311发芽率显著低的家系约有30%, 后熟21 d时比9311发芽率显著低的家系只有20%。
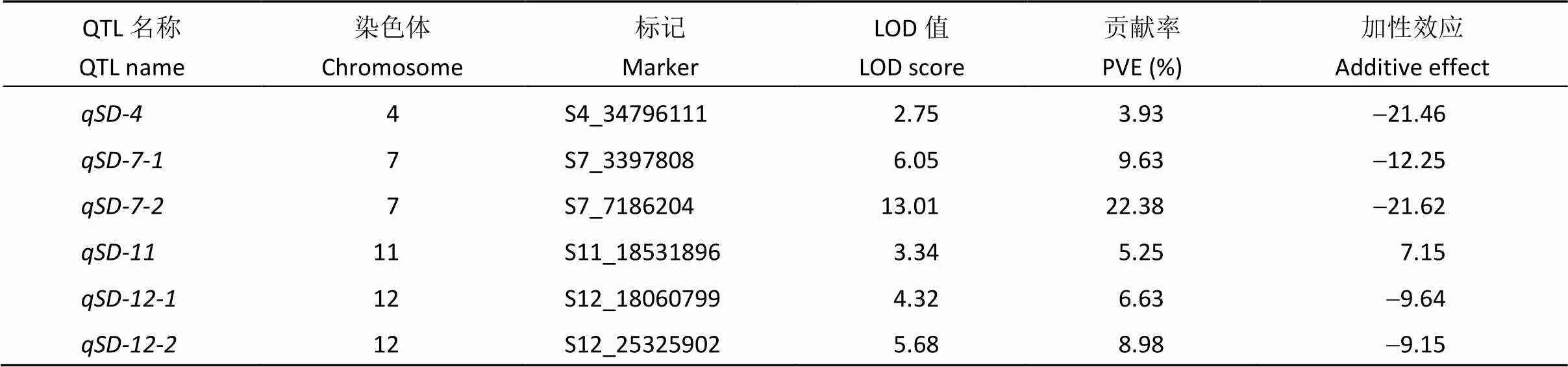
表2 2015年正季CSSL群体种子后熟21 d后休眠性QTL定位
PVE (%): 可解释的表型变异值。PVE (%): phenotypic variance explained。

表3 2015年正季CSSL群体种子后熟28 d后休眠性QTL定位
PVE (%): 可解释的表型变异值。PVE (%): phenotypic variance explained.
2.3 在CSSL群体中筛选休眠性强的家系
根据不同后熟时间后的种子休眠性表现, 选择了9个种子休眠性显著比背景亲本9311强的家系。2016年对所选家系的种子休眠性表型再次验证, 发现所有9个家系种子休眠性仍较强, 其发芽率均显著低于9311, 说明这些家系强的种子休眠性是能稳定遗传的。分析这些家系的基因型, 发现携带QTL位点越多的家系休眠性越强, 如在种子休眠性QTL位点携带多个野生稻置换片段的Q2、Q11、Q29、Q64等家系休眠性比仅带有单个或2个置换片段的家系休眠性更强, 表明这些QTL之间存在累加效应(表4和表5)。
2.4 利用Q14和9311衍生的F2次级分离群体验证qSD-7-2
2.4.1 F2群体休眠性表型分析 鉴于Q14在休眠性位点上携带的置换片段较少, 且已获得其与9311配制的F2群体, 为此, 2016年正季种植了Q14、9311及其由275个单株组成的Q14/9311的F2群体, 鉴定了后熟14 d后的种子发芽率, 尽管2016年收获时期连续阴雨天气, Q14与9311之间在种子休眠性上还是存在一定的差异, Q14的种子休眠性仍强于9311。Q14/9311衍生的F2群体的发芽率在0~90%间呈连续分布, 且群体表现偏向低发芽(强休眠性)的偏态分布。
2.4.2 验证对F2群体的休眠性QTL进行检测, 结果在第7染色体定位到2个QTL, 一个位于标记S7-11和S7-12之间, LOD值较小, 为2.88; 另一个位于标记RM180和RM21323之间, LOD值较高, 达18.49, 可解释表型变异的贡献率达33.53%, 为主效QTL, 该QTL位置与一致, 说明该位点能稳定遗传, 其来自普通野生稻的等位基因增强了种子休眠性(图4和表6)。
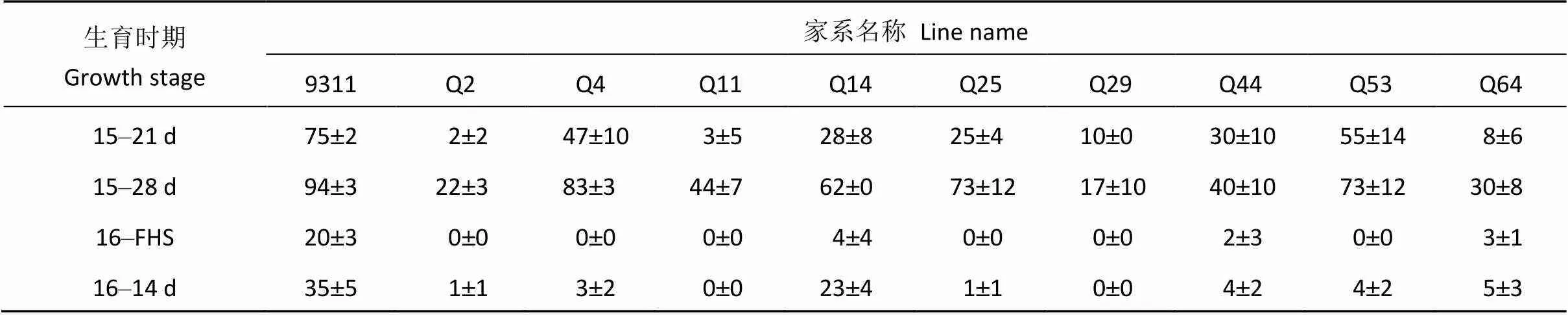
表4 CSSL群体中强休眠性家系的发芽率
15~21 d指2015年新鲜收获后熟21 d; 15~28 d指2015年新鲜收获后熟28 d; 16-FHS指2016年新鲜收获种子; 16~14 d指2016年新鲜收获后熟14 d; 发芽率表示为发芽率±标准差。
15–21 d: in 21 days after harvest in 2015; 15–28 d: in 28 days after harvest in 2015; 16-FHS: freshly harvested seeds in 2016; 16–14 d: in 14 days after harvest in 2016; Germination rate (%): germination rate ± standard deviation.

表5 CSSL群体中强休眠性家系的基因型
标记表示离检测到的休眠性QTL最近的标记; 空白表示该位点是9311基因型, “B”表示该位点是野生稻基因型, “H”表示该位点是杂合型。
Marker: the nearest marker from the tested QTL; blank: homozygous 9311 genotype; “B”: homozygous common wild rice genotype; “H”: heterozygous genotype.
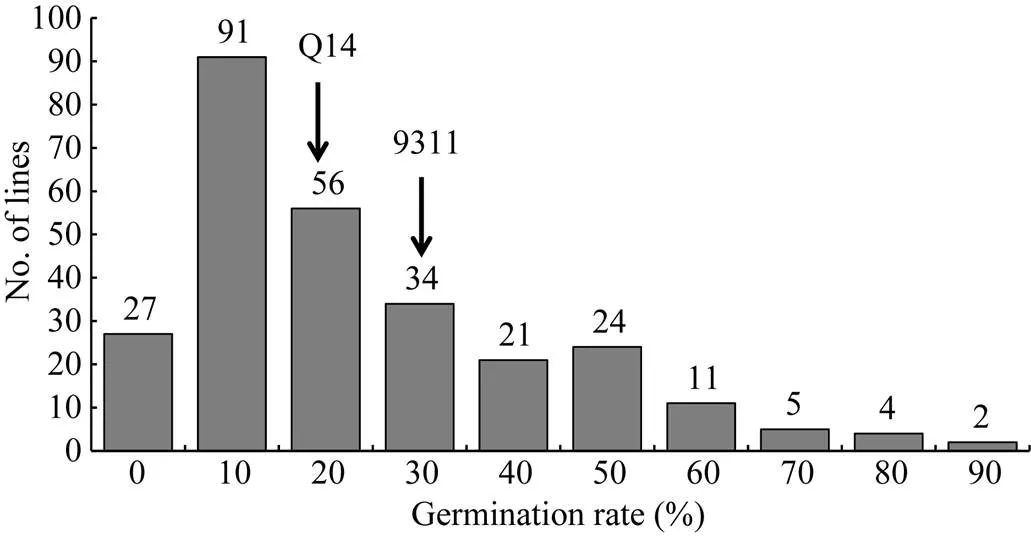
图3 2016年F2群体种子后熟14 d后休眠性次数分布图
Germination rate (%)表示种子在第7天的发芽率, 每个单株3个重复, 每个重复50粒; No. of lines为对应发芽率的单株数, 用柱子上的数字表示。
Germination rate (%) indicates that at seventh days of germination, each plant had three replicates to be tested, using 50 grains for each replicate; No. of lines in the number plants at the corresponding germination rate, showing on the column.
利用位点紧密连锁的分子标记RM180和RM21323分析Q14/9311 F2群体中各单株的基因型, 比较了2个标记同为9311型, 或同为普野型的单株的种子发芽率, 结果如表6,位点基因型为9311型的55个单株的平均发芽率为39%, 而为普野基因型的37个单株的平均发芽率只有11%, 两者存在极显著差异(表7), 说明来自普通野生稻的等位基因确实能降低种子发芽率, 增强种子休眠性。表明是真实存在的控制种子休眠性的位点。
3 讨论
野生稻是水稻育种的重要种质资源, 在其漫长的进化过程中形成了极其丰富的遗传多样性, 具有栽培稻所不具有或已消失了的优良基因。利用分子标记技术从野生稻中发掘有利基因是目前稻种资源研究利用中的重点。但其农艺性状往往不如栽培稻, 不良农艺性状和不利连锁基因的存在, 使对野生稻种质资源中优良基因的利用存在巨大挑战。1996年Tanksley等[16]首次验证了高代回交QTL分析法(advanced backcross quantitative traitlocus analysis, AB-QTL)的可行性, 此方法对野生种质资源中有利基因的研究和利用提供了新的途径。AB-QTL是利用高代回交群体分析QTL, 并且与轮回亲本不断回交构建近等基因系, 一方面验证所定位的QTL真实性, 另一方面可作为改良品系直接用于育种。CSSL是以AB-QTL分析法为基础, 通过杂交、连续回交和自交, 并结合分子标记辅助选择, 使供体亲本染色体片段渗入到受体亲本中。置换系的遗传背景与受体亲本相似, 只有少数置换片段的差异, 消除了背景干扰, 为遗传分析和功能研究奠定了良好基础。

表6 Q14×9311的F2群体中种子休眠性QTL定位
PVE(%): 可解释的表型变异值。PVE (%): phenotypic variance explained.

图4 Q14×9311的F2群体中种子休眠性QTL的LOD值曲线

表7 按qSD-7-2位点紧密连锁分子标记的基因型分析Q14/9311 F2群体各单株发芽率的株数分布
9311型: RM180/RM21323标记是纯合9311基因型; 普野型: RM180/RM21323标记是纯合普通野生稻基因型。
9311 type: RM180/RM21323 marker is homozygous 9311 genotype; Common wild type: RM180/RM21323 marker is homozygous common wild rice genotype.
现代高产品种往往缺乏种子休眠性, 在种子灌浆成熟后期遭遇高温多雨天气时, 极易出现穗发芽现象, 对作物生产造成严重损失。野生稻种子休眠性强, 因此可挖掘其休眠基因来改良常规品种。本研究利用普通野生稻和9311的染色体片段置换系为材料, 鉴定了控制普通野生稻种子休眠性QTL, 共定位到14个QTL, 分布在第3、第4、第5、第6、第7、第10、第11、第12染色体上。其中,在Miura等[18]利用日本晴/ Kasalath//日本晴的BIL群体检测到的区间内。位于Wang等[19]利用韭菜青和IR26的RIL群体检测到的区间内。Li等[20]利用珍汕97和明恢63的RIL群体在第5染色体上检测到位点, 本文的与其位置相近。Marzougui等[21]利用Nona Bokra (强休眠)和Koshihikari (弱休眠)衍生的染色体片段替代品系(CSSL)进行QTL分析。在第6染色体检测到一个QTL, 本研究检测到的与其位置相近, 与Gu等[22]利用EM93-1//EM93-1/SS18-2 (杂草稻株系)的BC1群体检测到的结果也相近。Sasaki等[23]利用日本晴和Kasalath的BIL群体在第10染色体检测到一个休眠性相关QTL, 本研究在相近位置同样检测到, 说明第10染色体该位置确实存在一个控制种子休眠的基因。本研究检测到的在后熟不同时间均检测到, 说明该位点不易被解除, 其位置与Rathi等[24]利用Cheni ahu (强休眠)和Kolong (无休眠)的F2群体检测的位置相近。此外, 本文检测到的的休眠性同样不易在后熟过程中解除, 且该位点在Cai等[17]利用台湾籼稻栽培品种和亚洲普通野生稻衍生的RIL群体检测到的区间内,即标记G148和Acp1之间。上述分析表明本研究检测到的QTL存在真实性。
本研究在第12染色体共检测到4个控制种子休眠性QTL, 其中和两个位点在后熟不同时间均被检测到, 说明二者在后熟过程中不易被解除。位于第12染色体短臂末端, LOD值为18.02, 可解释表型变异率为12.65, 加性效应为–11.90, 来自普通野生稻的基因增强种子休眠性, 目前在该位点尚未有相关报道, 可能是一个新的位点。此外,和两个位点距离很近且控制种子休眠性的基因来源不同, 如果这2个位点是真实存在的, 说明在普通野生稻的该区间存在作用相反的2个位点, 该结果还需要进一步验证。
本研究在后熟不同时间均检测到种子休眠性QTL, 说明该位点具有较强稳定性。进一步利用家系Q14与9311的F2群体验证第7染色体标记RM180和RM21323之间确实存在一个效应较大的QTL, LOD值为18.49, 可解释种子休眠性表型变异率为33.53%, 来自普通野生稻的等位基因显著增强种子休眠性。Gu等[22]利用杂草稻与栽培稻的BC1群体, 在后熟1 d、11 d、21 d均检测到第7染色体标记R180附近存在一个休眠性位点, 本研究的与其位置相近, 表明该位点可能普遍存在。但是该位点目前尚未被精细定位, 因此下一步计划是在家系Q14与9311的F2群体中挑选休眠性强的单株与9311继续回交, 进一步纯化遗传背景和缩小定位区间, 构建携带位点的近等基因系, 对其进行图位克隆。因此本研究不仅为水稻种子休眠基因的精细定位及克隆奠定了基础, 且为培育强休眠性籼稻品种提供了育种材料。
4 结论
定位到14个QTL, 分布在第3、第4、第5、第6、第7、第10、第11、第12染色体上。通过筛选强休眠性家系, 并分析这些家系所携带QTL数目, 发现家系携带的位点越多, 休眠性越强。利用家系Q14与9311的F2群体验证了位于第7染色体标记RM180和RM21323之间的QTL的真实稳定性, 该位点是一个控制普通野生稻种子休眠性的主效QTL。
[1] Bewley J D. Seed germination and dormancy., 1997, 9: 1055–1066
[2] Dong Y J, Tsuzuki E, Kamiunten H, Terao H, Lin D Z, Matsuo M, Zheng Y F. Identification of quantitative trait loci associated with pre-harvest sprouting resistance in rice (L.)., 2003, 81: 133–139
[3] 卢丙越. 水稻品种强休眠性的定位及遗传解析. 南京农业大学博士学位论文, 江苏南京, 2011 Lu B Y. QTL Mapping and genetic dissection of strong seed dormancy in N22 (L.). PhD Dissertation of Nanjing Agricultural University, Nanjing, China, 2011 (in Chinese with English abstract)
[4] Sugimoto K, Takeuchi Y, Ebana K, Miyao A, Hirochika H, Hara N, Ishiyama K, Kobayashi M, Ban Y, Hattori T, Yano M. Molecular cloning of, a regulator involved in seed dormancy and domestication of rice., 2010, 107: 5792–5797
[5] Takeuchi Y, Lin S Y, Sasaki T, Yano M. Fine linkage mapping enables dissection of closely linked quantitative trait loci for seed dormancy and heading in rice., 2003, 107: 1174–1180
[6] Gu X Y, Kianian S F, Hareland G A, Hoffer B L, Foley M E. Genetic analysis of adaptive syndromes interrelated with seed dormancy in weedy rice ()., 2005, 110: 1108–1118
[7] Gu X Y, Liu T L, Feng J H, Suttle J C, Gibbons J. Theunderlying gene promotes abscisic acid accumulation in early developing seeds to induce primary dormancy in rice., 2010, 73: 97–104
[8] Lu B Y, Xie K, Yang C Y, Wang S F, Liu X, Zhang L, Jiang L, Wan J M. Mapping two major effect grain dormancy QTL in rice., 2011, 28: 453–462
[9] 罗正良. 水稻抗穗发芽主效QTL的精细定位及候选基因分析. 四川农业大学硕士学位论文, 四川雅安, 2012 Luo Z L. Fine mapping and candidate gene analysis of, a major QTL for pre-harvest sprouting resistance in rice. MS Thesis of Sichuan Agricultural University, Ya’an, China, 2012 (in Chinese with English abstract)
[10] 钟代彬, 罗利军, 应存山. 野生稻有利基因转移研究进展. 中国水稻科学, 2000, 14: 103–106 Zhong D B, Luo L J, Ying C S. Advances on transferring elite gene from wild rice species into cultivated rice., 2000, 14: 103–106 (in Chinese with English abstract)
[11] Wan J M, Cao Y J, Wang C M, Ikehashi H. Quantitative trait loci associated with seed dormancy in rice., 2005, 45: 712–716
[12] Porebski S, Bailey L G, Baum B R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components., 1997, 15: 8–15
[13] Sanguinetti C J, Dias N E, Simpson A J. RAPD silver staining and recovery of PCR products separated on polyacrylamide gels., 1994, 17: 914–918
[14] Meng L, Li H H, Zhang L Y, Wang J K. QTL IciMapping Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations., 2015, 3: 269–283
[15] McCouch S R, Cho Y G, Yno M, Paul E, Blinstrub M, Morishima H, Kinoshita T. Report on QTL nomenclature., 1997, 14: 11-13
[16] Tanksley S D, Grandillo S, Fulton T M, Zamir D, Eshed Y, Petiard V, Lopez J, Beck-Bunn T. Advanced backcross QTL analysis in a cross between an elite processing line of tomato and its wild relative., 1996, 92: 213–224
[17] Cai H W, Morishima H. Genomic regions affecting seed shattering and seed dormancy in rice., 2000, 100: 840–846
[18] Miura K, Lin S, Yano M, Nagamine T. Mapping quantitative trait loci controlling seed longevity in rice (L.)., 2002, 104: 981–986
[19] Wang L, Cheng J, Lai Y Y, Du W L, Huang X, Wang Z F, Zhang H S. Identification of QTLs with additive, epistatic and QTL × development interaction effects for seed dormancy in rice., 2014, 239: 411–420
[20] Li W, Xu L, Bai X F, Xing Y Z. Quantitative trait loci for seed dormancy in rice., 2011, 178: 427–435
[21] Marzougui S, Sugimoto K, Yamanouchi U, Shimono M, Hoshino T, Hori K, Kobayashi M, Ishiyama K, Yano M. Mapping and characterization of seed dormancy QTLs using chromosome segment substitution lines in rice., 2012, 124: 893–902
[22] Gu X Y, Kianian S F, Foley M E. Multiple loci and epistases control genetic variation for seed dormancy in weedy rice ()., 2004, 166: 1503–1516
[23] Sasaki K, Kazama Y, Chae Y, Sato T. Confirmation of novel quantitative trait loci for seed dormancy at different ripening stages in rice., 2013, 20: 207–212
[24] Rathi S, Baruah A R, Chowdhury R K, Sarma R N. QTL analysis of seed dormancy in indigenous rice of Assam, India., 2011, 39: 137–146
Mapping of QTLs for Seed Dormancy inGriff.
SUN Ai-Ling1, WU Hong-Ming1, CHEN Gao-Ming1, ZHANG Tian-Yu1, CAO Peng-Hui1, LIU Shi-Jia1, JIANG Ling1,*, and WAN Jian-Min1,2
1State Key Laboratory of Crop Genetics and Germplasm Enhancement / Key Laboratory of Biology, Genetics and Breeding ofRice in Mid-lower Yangtze River, Ministry of Agriculture / Research Center of Jiangsu Plant Gene Engineering, Nanjing Agricultural University, Nanjing 210095, Jiangsu, China;2National Key Facility for Crop Gene Resources and Genetic Improvement / Institute of Crop Sciences, Chinese Academy of Agricultural Sciences, Beijing 100081, China
Seed dormancy of rice is an important agronomic trait related to rice quality and quantity. Studies on genetics and molecular mechanisms of rice seed dormancy are of great significance in breeding fine rice varieties with moderate dormancy. In this research, a set of chromosome segment substitution lines (CSSLs), derived from anrice variety 9311 as the recurrent parent and theGriff. as the donor parent, were used to detect the QTLs for dormancy of seeds at different storage dates after harvest. A total of 14 QTLs were detected on chromosomes 3, 4, 5, 6, 7, 10, 11, and 12. The lines with significantly stronger dormancy than the background parent 9311 were selected, showing the more dormancy loci in the lines the more strong dormancy. The F2population of the cross between Q14 and 9311 was used to verify the QTLs for seed dormancy. A significant dormancy locuswas mapped on chromosome 7 between the markers RM180 and RM21323, its LOD was 18.49 and the phenotypic variation rate was 33.53%. On this major stable inherited QTL, the allele gene fromGriff. significantly increased the dormancy of seeds. These results are available for map-based cloning of major QTLs for seed dormancy, and provide the breeding materials for cultivating appropriate dormant rice varieties.
Griff.; seed dormancy; CSSL; QTL
2017-04-06;
2017-09-10;
2017-10-31.
10.3724/SP.J.1006.2018.00015
通信作者(Corresponding author):江玲, E-mail: jiangling@njau.edu.cn
E-mail: 2014101098@njau.edu.cn
本研究由国家重点研发计划项目(2016YFD0100101-08), 江苏省农业科技自主创新资金课题(CX[16]1029), 安徽省科技重大专项(16030701068)和江苏省现代作物生产协同创新中心项目资助。
This study was supported by the National Key Research and Development Program of China (2016YFD0100101-08), the Agricultural Science and Technology Independent Innovation Fund Project of Jiangsu Province (CX[16]1029), the Science and Technology Major Project of Anhui Province (16030701068), and Jiangsu Collaborative Innovation Center for Modern Crop Production.
URL: http://kns.cnki.net/kcms/detail/11.1809.S.20171031.1427.002.html
猜你喜欢
防护林科技(2020年11期)2020-12-30 03:55:36
中国食品学报(2019年3期)2019-01-13 01:47:39
种子科技(2018年11期)2018-09-10 00:56:48
许昌学院学报(2018年8期)2018-09-05 02:05:18
蔬菜(2016年10期)2016-03-27 12:35:11
广东海洋大学学报(2015年4期)2016-01-13 08:39:30
天津农林科技(2015年1期)2015-12-30 13:05:14
听力学及言语疾病杂志(2015年5期)2015-12-24 01:47:04
首都医科大学学报(2015年4期)2015-12-16 13:00:08
武夷学院学报(2015年3期)2015-07-18 11:03:47
