
海水电池用Mg-Ga-Hg合金在模拟深海环境下的性能
2018-01-17 14:53:16徐海波王东鹏芦永红
电源技术 2017年12期
张 月,徐海波,王东鹏,芦永红
(中国海洋大学化学化工学院海洋化学理论与工程技术教育部重点实验室,山东青岛266100)
海水溶解氧电池(简称海水电池)是一种以金属材料为负极(活性溶解),海水为电解质,碳纤维或石墨等做正极(表面发生溶解氧还原反应)的化学电源[1]。海水电池最突出的特点是利用天然海水做电解质,能量密度高,敞开体系不需耐压装置,结构简单,安全性高,尤其适用于深海环境对小功率海洋观测设备进行长期供电[2]。
受海水介质(如温度、溶解氧浓度、海水流速等)的影响,易出现诸如正极氧还原活性低、负极金属溶解电流效率低且启动慢等问题,导致海水电池电能输出效果变差[3-4]。本课题组研制的第一代海水超级电容溶解氧电池[5](SWB-EC)实海测试时发现,采用的Mg-Mn合金负极极化严重、滞后效应明显,从而使材料的利用率降低,电池的输出电压和体积功率密度较低且影响启动。YU等[6]研究发现,Mg-Ga-Hg合金在20 mA/cm2的电流密度放电时电极电位仍可保持在-1.8 V且易启动。高春燕等[7]将Mg-Ga-Hg合金用于SWB-EC,发现在不同盐度的海域中小电流密度(1~9 mA/cm2)下工作时,开路电位可低至-1.91~-1.93 V(vs.SCE),能迅速激活且间歇放电好,电流效率高,作为海水电池负极材料具有潜在的应用价值,但目前尚缺乏深海环境中的测试及评价。无疑,深海中的低温、低流速等恶劣条件会增加海水电池的供电难度。因此,针对深海使用工况选择高性能的海水电池负极材料就显得十分重要。
本文在模拟深海环境条件下,评价了不同温度和流速下Mg-Ga-Hg合金出厂态和间歇工作过程中的电化学性能,探讨了Mg-Ga-Hg合金用作深海海水电池负极的可行性。
1 实验
1.1 实验装置及电化学测试
采用三电极体系:饱和甘汞电极(SCE)作参比电极、钛基氧化铱(DSA)电极作辅助电极,1 cm2工作面的Mg-Ga-Hg合金(Mg-2%Ga-3%Hg)做工作电极,非工作面用环氧树脂密封,电解液为青岛天然海水。动态条件下的测试实验装置见图1,用直流潜水泵使海水在储液池和反应池之间循环来模拟海流,控制海水流速为4 cm/s。通过YT-DW-02低温恒温循环水浴控制海水温度,电化学测试使用ZF-9恒电位/恒电流仪,文中电位均相对于饱和甘汞电极。

图1 动态条件下电化学测试装置
主要电化学测试包括开路电位和恒电流实验。测试前试样经600#、1000#、1200#水磨砂纸逐级打磨,用蒸馏水清洗,热风吹干。为模拟海水电池运行过程中镁棒消耗引起的电流密度变化,恒电流测试时电流密度分别为1、3、6、9 mA/cm2。为模拟海水电池出厂态和间歇工作过程,恒电流测试分为将商品合金试样直接在海水中电解和在海水中浸泡1 h后再进行电解两种。
1.2 自放电速率测试
采用失重法测定镁合金自放电速率:试样尺寸为18 mm×18 mm×0.28 mm,准确测出初始质量m0和面积S,打孔,用细线将其系住,采取悬挂的方式将四个平行试样完全浸泡于500 mL海水中,避免试样与烧杯底部接触,24 h后取出清洗,再放入200 g/L铬酸(CrO3)溶液中浸泡10 min,取出后用蒸馏水清洗、烘干,记录剩余质量m1。自放电速率用电流密度表示,按式(1)计算:
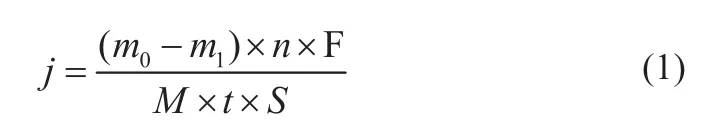
式中:j为自放电速率,mA/cm2;m为质量,mg;t为浸泡时间,s;S为表面积,cm2;F 为法拉第常数,96 500 C/mol;M为镁的摩尔质量,24 g/mol;n为镁失电子数,为2。
1.3 电流效率计算
准确测出处理好的工作电极的初始质量m0'和面积S',在不同的电流密度j'下进行放电,通过控制不同电流密度下的电解时间保证每次电解时电量都为13 500 C,放电结束后用蒸馏水清洗,放入200 g/L的CrO3溶液中浸泡10 min,取出后用蒸馏水清洗、热风吹干,记录剩余质量m1'。电流效率η按式(2)计算,即按纯镁计算的理论质量损失与实际质量损失的比值,电流效率是三个平行样的平均值。

式中:j'为电流密度,mA/cm2;m0'为初始质量,mg;m1'为放电后质量,mg;t'为放电时间,s;S'为表面积,cm2;F 为法拉第常数,96 500 C/mol;M为镁的摩尔质量,24 g/mol;n为镁失电子数,为 2。
2 结果与讨论
2.1 海水温度和流速对Mg-Ga-Hg合金开路电位的影响
图2是Mg-Ga-Hg合金在不同温度和流速的海水中12 h内的开路电位-时间曲线,可以看出,不同工况下2 min内Mg-Ga-Hg合金的开路电位即可稳定在-1.92 V以下(见放大图)。较低开路电位有利于增加电池的输出电压,使其具备高活性负极材料的基本要求。另外,相较而言,20℃时静/动态条件下Mg-Ga-Hg合金开路电位相差不大,而20与4℃时差别略大,可见流速对开路电位的影响不及温度。有利的是,Mg-Ga-Hg合金在4℃低温动态海水中的开路电位可低至-1.95 V,初步判断深海环境的低温特点对Mg-Ga-Hg合金负极性能是有利的。若将其与本课题组研制的改性PAN碳纤维刷正极材料组装成SWB-EC,理论输出电压将可达1.50 V以上。图中各曲线均存在少许的电位波动,源于腐蚀产物在镁基体表面的堆积-溶解平衡。腐蚀产物先沉积在镁合金表面形成保护膜,开路电位正移;而镁合金中添加的Ga和Hg易于形成镁汞齐,又使金属不断活性溶解带离腐蚀产物[8],开路电位负移,这一动态平衡可使电位基本保持稳定。

图2 Mg-Ga-Hg合金在不同温度和流速海水中的开路电位-时间曲线
2.2 海水温度和流速对自放电速率的影响
自放电速率是影响镁合金海水电池负极利用率的重要因素,表1是Mg-Ga-Hg合金在不同温度和流速海水中24 h内的自放电速率比较。添加了高析氢过电位元素Ga和Hg后,Mg-Ga-Hg合金的析氢反应活化能增大,使腐蚀原电池导致的溶解过程被阻滞[9]。试样在20℃、静止海水中的腐蚀速率仅为0.157 mA/cm2;流速增加到4 cm/s,腐蚀速率增加到3倍以上,说明流速对其影响较大。镁合金发生析氢[式(3)]的初期反应速率较快,随着反应进行,沉积到表面的腐蚀产物Mg(OH)2累积形成保护膜,一定程度上抑制了腐蚀的进一步发生,故静态时自放电速率较低。动态时腐蚀产物膜会随着海水流动不断脱落,使新鲜基体裸露并不断溶解,自放电速率增加。降低温度(4℃)后,析氢反应速率随之减小,自放电速率降为原先的2/3,可见深海低温环境(4℃)利于自放电速率的降低和镁合金利用率的提高。

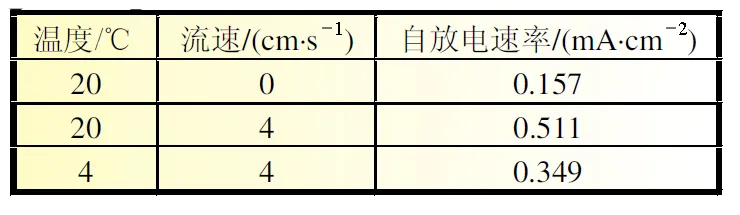
表1 Mg-Ga-Hg合金在不同温度和流速海水中的自放电速率
2.3 海水流速和温度对Mg-Ga-Hg合金电流效率的影响
海水流速和温度对Mg-Ga-Hg合金放电电流效率的影响如 表2所 示 ,η1、η3、η6、η9分别表示电流密度为1、3、6、9 mA/cm2时镁合金的电流效率。由表2可知,静态时镁合金的电流效率要高于动态时,且随电流密度增加电流效率提高。根据表1自放电速率的变化,与动态相比,静态时腐蚀产物的阻碍会导致自放电速率降低和电流效率升高;增大施加电流,镁合金用于电池放电的比重增加,导致电流效率大致呈上升趋势。动态时,海水流动减小了腐蚀产物的影响,使自放电速率增加、放电电流效率有所降低;增加外加电流的强制溶解,有效溶解加剧和自放电速率的增加相互抵消,使电流效率对电流密度的变化不敏感而保持基本不变。温度降低,自放电速率的降低利于电流效率的提高,使4℃动态海水中的电流效率比20℃时高0.4%~2.2%,也表明深海环境的低温特点对Mg-Ga-Hg合金负极性能是有利的。不同工作电流密度下的电流效率均可达50%以上且波动不大,可以判断随着Mg-Ga-Hg合金负极不断消耗,在使用后期表面积减少引起的电流密度增大也不会显著影响负极的放电性能。

表2 海水流速和温度对Mg-Ga-Hg合金放电电流效率的影响
2.4 Mg-Ga-Hg合金的恒流放电性能
电压滞后是指恒流放电开始时,由于腐蚀、钝化等原因使电极电位先变正(对应的电位称为滞后电位)然后负移,经过一定时间(从起始到电位稳定经过的时间称为激活时间)后才能达到稳定放电电位(即工作电位)的现象[10]。通常滞后电位越负、激活时间越短,海水电池的启动性能越好。除材料自身性质外,负极的工作电流密度也是其主要影响因素。图3比较了1~9 mA/cm2下出厂态Mg-Ga-Hg合金的放电性能,放大图显示激活时间,滞后电位用箭头标出。总体而言,随电流密度增加,滞后电位变正,滞后效应明显,激活时间增加,稳定工作电位变正,具体参数见表3。这说明初期较大电流启动和后期电极面积减小都会使启动变得困难。由表3可知,静态海水中,随电流密度增加,滞后电位变正,激活时间更长,电池启动性能下降,大电流启动变得更难。流速为4 cm/s时,激活时间大大缩短,在10 s内工作电位均能达到-1.90 V以下,且1、3、6、9 mA/cm2电流密度下稳定工作电位较静态分别负移34、40、73和81 mV,说明流动条件不仅有利于电池启动,也有助于电池稳定工作时输出电压的提高。对比低温(4℃、4 cm/s)条件,除小电流密度外,温度对滞后电位和工作电位的影响不大而对激活时间的影响较大。然而,尽管激活时间有所增加(尤其是大电流密度时),但仍大大低于静态。因此,只要保证流速,在低温下海水电池仍具有良好的启动性能。
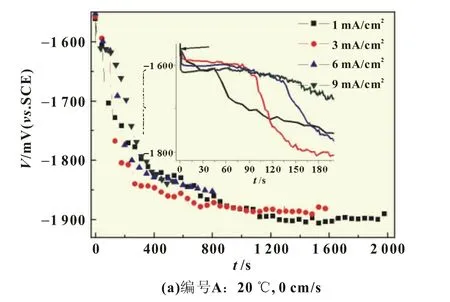

图3 不同电流密度下Mg-Ga-Hg合金在海水中的恒流放电曲线

表3 Mg-Ga-Hg合金的恒流放电性能参数
2.5 Mg-Ga-Hg合金间歇工作时的恒流放电性能

图4 不同电流密度下Mg-Ga-Hg合金在海水中浸泡1 h后的恒流放电曲线

表4 Mg-Ga-Hg合金在海水中浸泡1 h后放电性能参数
为模拟海水电池间歇工作(即停止工作,然后再次启动)时电流密度对材料放电性能的影响,将Mg-Ga-Hg合金在海水中自然浸泡1 h后在1~9 mA/cm2电流密度下进行恒流放电,测试结果如图4,相关参数见表4,放大图显示激活时间,滞后电位用箭头标出。由图4可知:浸泡后相对活化的镁合金再次启动后的放电性能与出厂态相似,即随电流密度的增加滞后电位正移,激活时间增加,稳定工作电位正移。对比表3,表4中静态条件下Mg-Ga-Hg合金的滞后电位正移幅度较大,但激活时间减少约一半,稳定工作电位负移,这说明合金表面的腐蚀产物和出厂态自然氧化膜对电池启动的影响不同。于4 cm/s的动态海水中再次启动时腐蚀产物的影响基本消除,滞后电位明显负移,并能在较短时间(1 min)内激活,稳定工作电位负移且保持在-1.90 V以下。低温时滞后电位稍正,但激活时间和稳定工作电位几乎不变。这说明海水电池间歇工作再次启动时,海水流速能显著消除腐蚀产物的影响,保持其良好的启动性能,而低温对启动过程的不利影响极易消除,仍能保证海水电池的快速启动和稳定的放电性能。
实际应用中,电池安装需要一定的时间,在此条件下,电池浸泡后即可快速启动,这是非常利于电池正常工作的。使用时间后,即使电池短时间内停止工作,再次启动也会非常,深海低温环境对其影响不大,均可快速达到正常输出电证明了Mg-Ga-Hg合金在深海环境中优良的综合性能。
3 结论
在模拟深海低温动态环境(4℃,4 cm/s)中,Mg-Ga-Hg合开路电位低于-1.95 V,作为海水电池负极材料在1~9 cm2电流密度下放电时,稳定工作电位仍低于-1.90 V,且的自放电速率可使电流效率维持在50%以上。此外,对出厂态和间歇工作状态,Mg-Ga-Hg合金的滞后电位负、激活时间短,均可快速启动。故此,Mg-Ga-Hg合金有望用作海水电池的负极材料以满足深海观测仪器长期供电需求。
[1]HASVOLD Ø,LIAN T,HAAKAAS E,et al.CLIPPER:a long-range,autonomous underwater vehicle using magnesium fuel and oxygen from the sea[J].Journal of Power Sources,2004,136(2):232-239.
[2]王宇轩,黄雯,李学海.水下电源的研究进展[J].电源技术,2011(7):858-861.
[3]NARAYANAN S R,SATHYANARAYANA S.Voltage delay during constant-currentor constant-resistance discharge of Mg-MnO2dry cells:a comparative study[J].Journal of Applied Electrochemistry,1989,19(4):495-499.
[4]PARDO A,MERINO M C,COY A E,et al.Influence of microstructure and composition on the corrosion behaviour of Mg/Al alloys in chloride media[J].Electrochimica Acta,2008,53(27):7890-7902.
[5]徐海波,芦永红,张伟,等.海水超级电容溶解氧电池[J].电化学,2012(1):24-30.
[6]YU K,TAN X,HU Y,et al.Microstructure effects on the electrochemical corrosion properties of Mg-4.1%Ga-2.2%Hg alloy as the anode for seawater-activated batteries[J].Corrosion Science,2011,53(5):2035-2040.
[7]高春燕,芦永红,徐海波.海水电池用Mg-Ga-Hg合金的电化学性能[J].中国海洋大学学报:自然科学版,2015,45(10):69-73.
[8]FENG Y,WANG R,YU K,et al.Influence of Ga and Hg on microstructure and electrochemical corrosion behavior of Mg alloy anode materials[J].Transactions of Nonferrous Metals Society of China,2007,17(6):1363-1366.
[9]YAN F,WANG R,PENG C,et al.Influence of Mg21Ga5Hg3compound on electrochemical properties of Mg-5%Hg-5%Ga alloy[J].Transactions of Nonferrous Metals Society of China,2009,19(1):154-159.
[10]宋光铃.镁合金腐蚀与防护[M].北京:化学工业出版社,2006:41-42.
猜你喜欢
小读者·爱读写(2023年9期)2023-10-02 03:46:59
小读者(2023年18期)2023-09-27 04:38:38
湿法冶金(2020年1期)2020-02-24 06:22:04
中国有色金属学报(2018年2期)2018-03-26 07:58:37
科学与财富(2017年9期)2017-06-09 18:45:34
中国氯碱(2017年3期)2017-04-18 02:23:04
电镀与环保(2016年3期)2017-01-20 08:15:29
电源技术(2016年6期)2016-04-05 08:46:30
电源技术(2015年7期)2015-08-22 08:48:40
电源技术(2015年9期)2015-06-05 09:36:06
