
极长与极短的铜铜距离-吡唑环三核亚铜配合物亲铜作用本质及发光调控
2018-01-12 05:57邢立锐
物理化学学报 2017年11期
郑 霁 邢立锐 李 丹,*
极长与极短的铜铜距离-吡唑环三核亚铜配合物亲铜作用本质及发光调控
郑 霁1邢立锐2李 丹1,*
(1暨南大学化学与材料学院,广州 510632;2汕头大学化学系,广东 汕头 515063)
本文结合近年来亲金属作用研究领域的进展,针对本课题组在环三核亚铜配合物方面的最新研究成果,讨论了两个铜(I)–铜(I)作用相差悬殊的体系。一是通过构筑具有环三核单元的三棱柱笼状配合物,确认在正堆积模式下即使环三核单元之间亲铜作用极弱依然可以在磷光发射态中产生强亲铜作用,且通过配体的预留配位点与Cu2I2簇连结从而得到自校准大范围发光温度计;二是通过与亲铜作用正交的Br―Br卤键,实现环三核亚铜配合物前所未有的极短铜(I)–铜(I)距离,通过各种电子结构分析方法研究其本质。结果表明即使当铜(I)–铜(I)距离很接近铜的范德华半径和时,其本质依然为闭壳层作用,而Br―Br作用总为闭壳层作用,且该体系中最强的Br―Br作用很好地体现出一个Br原子的穴和另一个Br原子的负静电势区域的匹配性。
亲铜作用;正堆积;磷光发射态;卤键;约化密度梯度;分子中的原子的量子理论
1 引言
电子组态为10的铸币金属一价阳离子之间的金属-金属作用(即亲金属作用)逐渐成为一种被广泛用于超分子聚集体构筑及晶体工程设计的超分子作用,在发光、催化等领域得到持续的研究及应用1–5,然而,这种弱作用的本质一直存在争议6–10。近年来,亲金属作用研究中的争论主要集中在基态和激发态中这种作用是否总是同时存在或同时不存在,以及基态和激发态中其本质是否不同。
一般而言,假如基态存在(或不存在)亲金属作用,那么激发态中往往也存在(或不存在)亲金属作用。然而,这种情况并非必然成立。例如,3,5-二(三氟甲基)吡唑的环三核亚铜配合物的晶体中,最短分子间铜(I)–铜(I)距离已经长达0.3879 nm (298 K)11和0.3813 nm (100 K)12,远超过铜(I)的范德华半径和(0.28 nm),故Vorontsov13认为此晶体不存在基态的分子间铜(I)–铜(I)作用。然而,该晶体在不同温度下皆呈现出此类体系特征性的亲铜作用主导的低能红、橙光。根据其激发态单晶结构测试13以及Hu14的理论计算结果,发现该配合物在磷光发射态中存在显著的分子间亲金属作用。这是基态不存在亲金属作用而激发态存在亲金属作用的一个典型的例子。
目前一般认为,基态的10金属离子之间的亲金属作用为色散力主导,而激发态的则更多地体现为共价键。随着科技的发展,与激发态结构相关的测试报道也逐渐增多。如近年来发表的通过飞秒X射线散射技术对Au(I)配合物的研究15和时间分辨Laue衍射技术对混Ag/Cu配合物的研究16,都明确观察到激发态中形成金属–金属共价键的现象,而其基态时的亲金属作用都基本被归结为色散力。
作为铸币金属离子的环三核配合物发光领域的先驱,Vorontsov等13在2005年的激发态单晶结构测试,以及Grimes17、Hu14等在2006–2007年详尽的理论计算确定了环三核配合物的低能磷光由分子间亲金属作用主导,从此奠定了环三核体系磷光研究的基调,即围绕着分子间亲金属作用的调控。基态与激发态的多重亲金属作用位点及与之相关丰富的磷光行为,令环三核体系成为很适合研究亲金属作用构效关系的一类典型体系。在基于过渡金属配合物的磷光材料方面,亲金属作用的存在往往有利于提高系间穿越(ISC)效率、缩短辐射跃迁寿命,减少非辐射跃迁对能量的损耗,故常可提高磷光量子产率18。
铸币金属的吡唑基环三核配合物的合成及构效关系研究,一直是我们课题组的一个主要研究兴趣19–28,其中我们最关注的是其丰富的发光性质的调控。本文主要围绕本课题组近年来在环三核亚铜配合物方面的部分成果,结合目前在亲金属作用研究领域在基态和激发态方面的争议进行总结,希望对投身本领域研究的科研工作者提供一点启发与借鉴。
本文主要聚焦于两类与亲金属作用相关的例子,一是通过配体设计,得到具有双环三核单元的六核笼状配合物,实现两个环三核单元正堆积模式,在基态环间亲金属作用很弱的前提下,依然在磷光发射态中产生显著的亲金属作用;二是通过与亲金属作用正交的Br―Br卤键,形成环三核亚铜配合物领域中未曾报道过的极短的分子间铜(I)–铜(I)距离,通过一系列电子结构分析方法,对铜(I)–铜(I)作用和卤键的本质进行较深入的分析。
2 具有双环三核基元的六核配合物
环三核亚铜配合物常以错位堆积和滑移堆积的方式形成晶体,有时也采用椅式堆积的模式,后者可看作前两者的组合(图1)。然而,截至2017年5月,环三核亚铜配合物的晶体结构报道中都没有出现完全面对面的正堆积模式的报道,其原因是这种堆积方式下相邻分子间带有同号静电势的区域直接正对,从而产生很大的静电排斥,且此模式下位阻也较大。即使在正堆积模式下可以形成三对分子间亲铜作用,也可能不足以克服这种堆积方式下伴随的强库仑排斥。
2011年,我们报道了首个环三核亚铜单元以正堆积形式存在的例子23。通过设计合成由二亚甲基苯基连结两个3,5-二甲基吡唑基团的半刚性配体,与Cu2O反应成功得到以两个环三核单元组成的六核三棱柱笼状亚铜配合物(图2)。随后,我们将配体换成2,6-二甲基-3,5-二亚甲基吡啶基,得到另一例同构的亚铜化合物24。比较这些双环三核正堆积的笼状亚铜配合物和已见报道的单环三核亚铜寡聚物,我们发现以下有趣的特点:
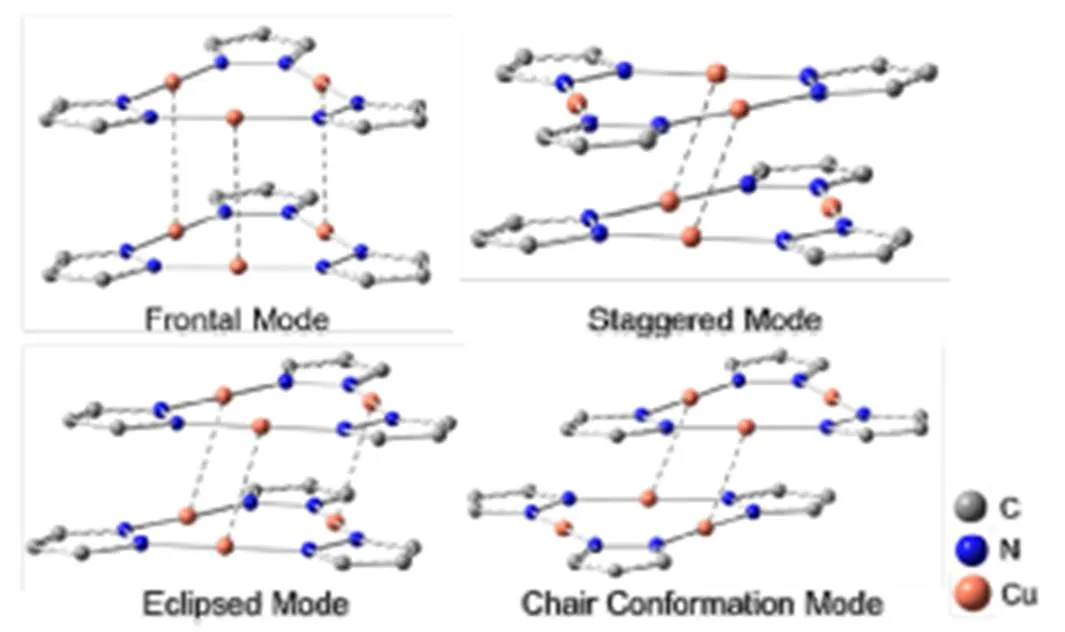
图1 吡唑基双环三核亚铜配合物的四种代表性堆积模式
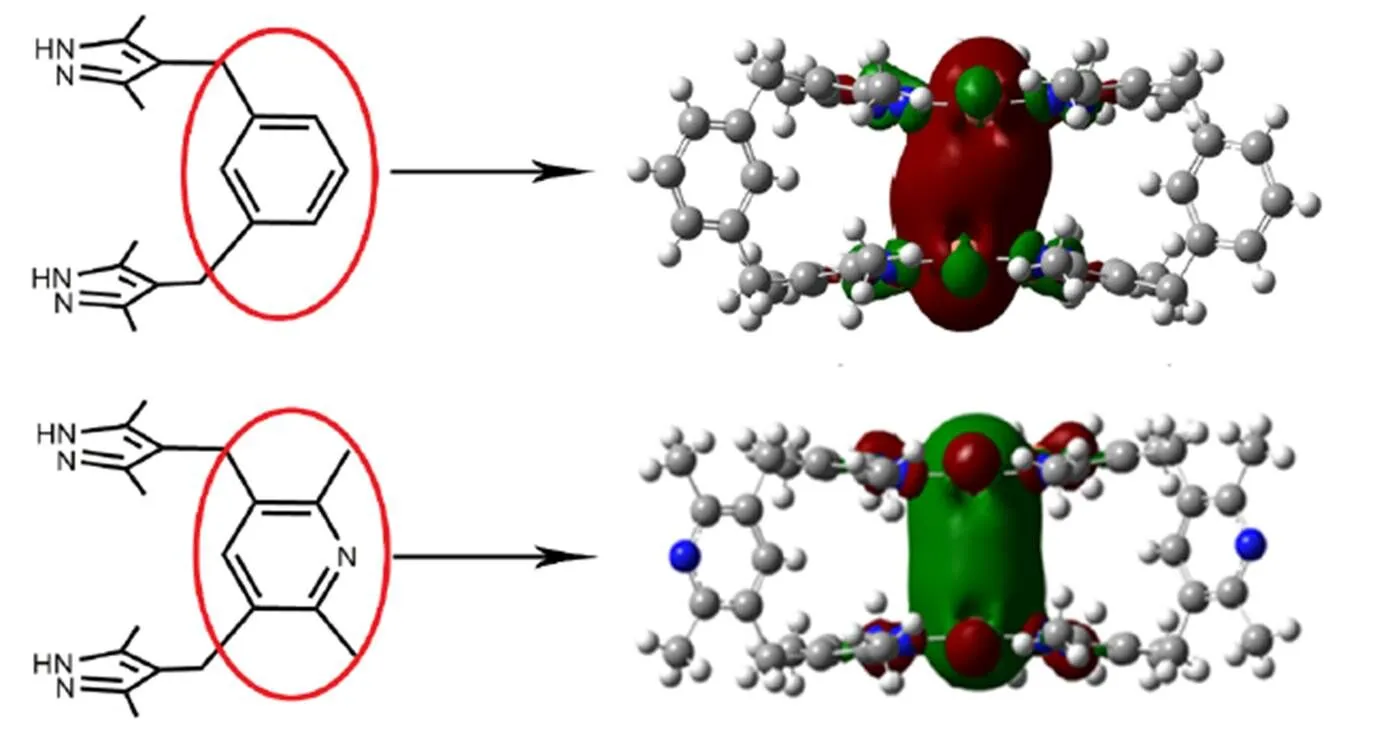
图2 用红圈凸显不同连接体的联吡唑配体(左)及其对应的笼状配合物的最低空轨道(右)
一、尽管从基态单晶结构来看,这两例六核化合物的分子内环三核单元间铜(I)–铜(I)作用都很弱,含苯基的例子中,铜(I)–铜(I)距离在0.370–0.395 nm范围内,含吡啶基的例子中,最短的铜(I)–铜(I)距离已经超过0.400 nm,此时已不能认为存在基态的环三核单元间亲铜作用。然而,这些笼状化合物都展现环三核体系亲金属作用连结的激基缔合物的特征低能磷光。笼状化合物晶体堆积结构中,来自两个相邻笼的环三核间最短铜(I)–铜(I)距离总是比笼内的环三核单元间的大0.01 nm以上,因此,发光可能源于激发态的笼状分子内环三核单元间亲铜作用。
二、从发光行为来看,与环三核亚铜寡聚物相比,六核笼状配合物的固态磷光的最大发射波长在各种实验测定温度下基本都超过710 nm,具有巨大的斯托克位移,而已报道的环三核寡聚物晶体的磷光最大发射波长都未超过670 nm。并且,对于含苯基的六核笼状配合物,其磷光最大发射峰在常温到10 K的范围内并不发生明显位移,也不会出现高能肩峰,类似的现象也发生在含吡啶基的六核笼状配合物的晶体中,即不展现出明显的光致发光变色现象,这与经典的环三核体系明显不同。如此与经典环三核寡聚物迥异的磷光性质,进一步支持我们上面的推测,即我们的笼状配合物的晶态磷光很可能源于激发态的笼状分子内环三核单元间亲铜作用,而不是晶体中两个相邻笼状配合物的分子间亲铜作用。毕竟,笼状配合物之间即使存在亲铜作用,也是配体不支持的,实质上与经典的环三核寡聚物的分子间亲金属作用没有本质区别。
三、对这两例笼状配合物模型进行密度泛函理论计算,发现尽管环三核单元间距离甚远,其基态的最低空轨道(LUMO)都是典型的体现环三核单元间亲金属作用成键性质的轨道,这与经典的吡唑环三核体系一致,此类轨道在环三核体系中对亲金属作用主导的磷光发射发挥最重要的作用。该类体系常表现出的在最低三重态下铜(I)–铜(I)作用被增强的现象,主要是源于电子从Cu(I)离子的3轨道跃迁到4/杂化轨道,令LUMO被电子布居,从而体现出此轨道的成键性质,提高铜原子的成键能力,因此导致磷光发射态中环三核单元间铜(I)–铜(I)距离缩短,体现出光诱导Jahn-Teller扭曲的现象。
以上特点,说明通过有机连接体强制连结而得到的正堆积模式下,尽管基态的亲铜作用很弱(或甚至不存在),但磷光发射态下依然足以形成较强的环三核单元间亲铜作用。并且,尽管都是环三核单元间亲铜作用主导的磷光,这类笼状配合物的发光却又呈现出不同于经典环三核寡聚物的特征。
由环三核亚铜单元间亲铜作用主导的磷光,光谱常表现为黄光到红光区的发射宽峰。该单元体系已经成为一类经典的发光团。在此基础上,我们使用带吡啶基的吡唑衍生物作为配体,利用CuI引入另一类经典的发光团铜碘簇29,成功将吡唑环三核单元与铜碘簇结合,制备出一系列吡唑环三核亚铜单元与Cu3I3簇25或Cu4I4簇26共存的两例配位聚合物。这两例配位聚合物发射光谱均保留了结构中两个经典发光团独立的发射光特性,呈现双发射峰光谱曲线,而且,在外界刺激(如温度)下,两个发射峰变化的程度不一,导致化合物宏观的发光性质随外界刺激不同而不同,这一策略被我们命名为“化学调色板(Chemopalette)”,即通过分子中存在的两个独立发光团来实现对分子整体发光性能的有效调控。这两例报道中,与铜碘簇相关的磷光能量都较低,其磷光带与吡唑环三核发光团的能量比较接近,从而令整体发光依然偏低能,难以调配出蓝色或紫色等颜色的发光。
我们使用上文中提及的以2,6-二甲基-3,5-二亚甲基吡啶基作为连接体的六核笼状配合物与碘化亚铜反应,利用配体上预留的未配位的吡啶氮原子与Cu2I2簇配位,得到通过一个Cu2I2簇连结两个六核配合物的十四核配合物24。
在这个配合物中,预先合成的六核笼状配合物保证了环三核单元间的铜(I)–铜(I)作用的数量及距离被控制在一定的范围内;吡啶氮原子的两个邻位各被一个甲基占据,位阻导致此氮原子只能与Cu2I2簇(而非更高核数的铜碘簇)配位,而Cu2I2簇配合物的磷光基本都体现为从卤素和铜原子到有机配体的电荷转移,其发光颜色几乎涵盖整个可见光区29,30。另外,此十四核配合物的环三核与铜碘簇部分并不以共轭化学键相连,使这两部分的磷光发射态之间的耦合程度很低,阻碍它们之间的内转换。在不同温度下,此配合物的晶态样品的发射光谱都保持着明显的高能峰(~430 nm)与低能峰(700–730 nm) (图3(a, b))。通过激发波长和温度的调控,可展现出蓝光和紫光发射,这无疑是又一个典型的化学调色板的例子。并且,我们很幸运地发现此配合物可设计为宽范围(从100到450 K)的自校准发光温度计(图3(c))。通过与六核笼状配合物及2,5-二甲基吡啶的Cu2I2配合物进行发光性质、分子轨道的比较,发现十四核配合物的发射谱及电子结构基本接近于这两种低核配合物的简单叠加。
3 与卤键正交的分子间铜(I)–铜(I)作用
以3,5-二甲基-4-(4′-溴)苯基吡唑为配体,在溶剂热条件下得到环三核亚铜配合物的一对真正的超分子异构体及其赝超分子异构体(环三核配合物与对二甲苯的共晶),这三种晶体皆采取椅式二聚体的堆积模式。其中一种异构体(命名为Br–α)的分子间铜(I)–铜(I)距离为0.2859 nm (293 K)和0.2817 nm (100 K),非常接近铜原子的范德华半径和(0.28 nm),且分子本身明显扭曲,构成二聚体的两个分子明显外翻,令二聚体呈现双叶双曲面的形状(图4)28。Vorontsov等13早已于2005年指出,在吡唑环上引入拉电子取代基,会导致环三核配合物倾向于以铜(I)–铜(I)距离较远的一维折叠链形式堆积;相反,引入推电子取代基则会导致倾向于以铜(I)–铜(I)距离较近的二聚体形式堆积。但到目前为止,只有3,5-二甲基吡唑13、3,5-二异丙基吡唑13及3-(2’-吡啶基)吡唑31的环三核亚铜配合物晶体展现出短于0.300 nm的分子间铜(I)–铜(I)距离,但都长于0.290 nm。我们的这种溴基配合物形成的三种(赝)超分子异构体中,最短分子间铜(I)–铜(I)距离的分布范围很广,分别为约0.28、0.30和0.32 nm,说明此体系中极短的铜(I)–铜(I)距离很可能不是由于取代基的推电子效应,而与特定晶体堆积方式中的超分子作用力相关。为了更深入地揭示其本质,我们对这些晶体进行了一系列基于电子密度的理论计算分析。
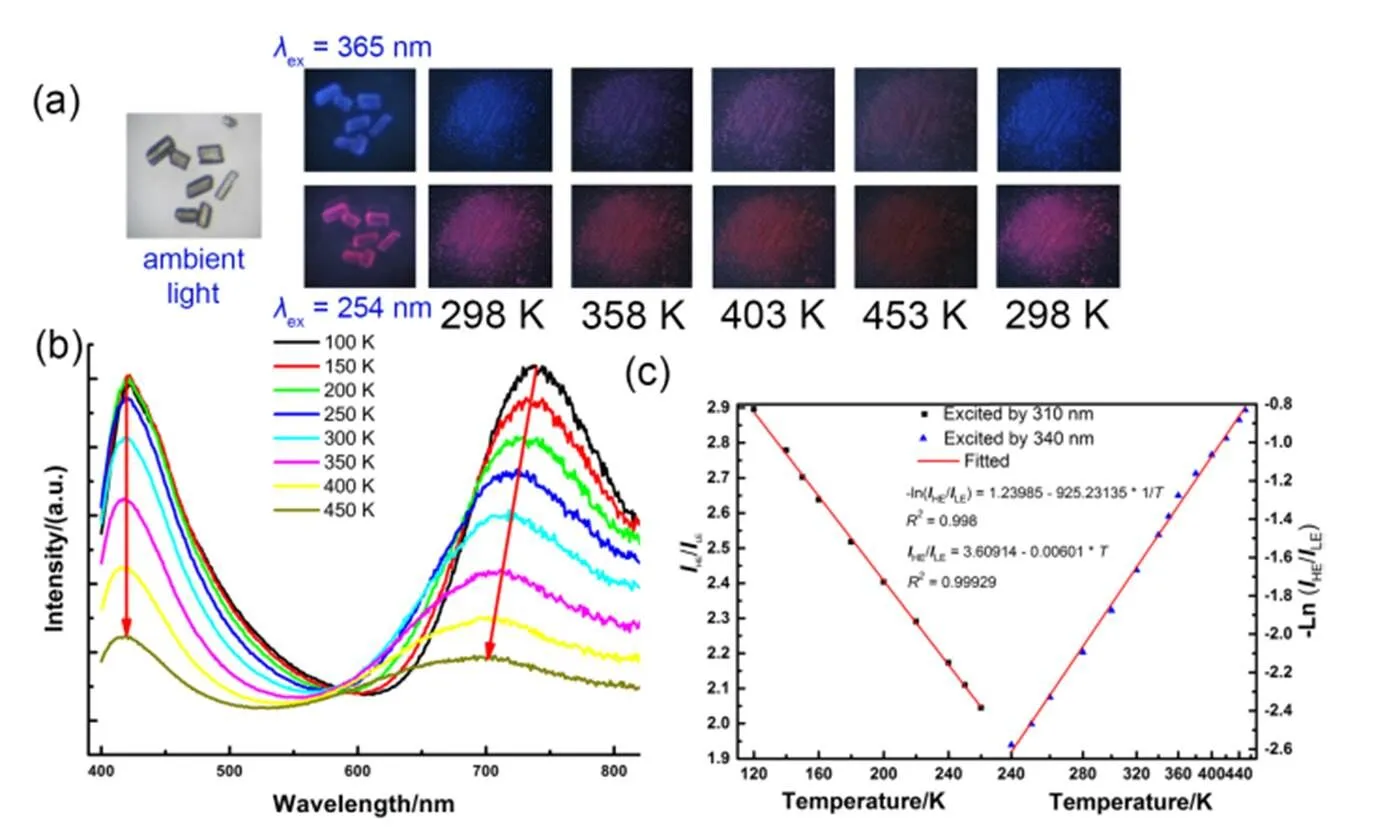
图3 (a)十四核配合物的晶体在不同温度及不同激发波长的紫外灯下的发光照片、 (b)290 nm激发下的发射光谱(箭头表示最大发射波长处的强度变化)及(c)工作曲线24
Fig.3 (a) Visual temperature monitor of the tetradecanuclear complex under UV lamp with differentex; (b) temperature-varied emission spectra excited at 290 nm (Arrows indicate the intensity changes atemmax); (c) Working curves24.
通过Hirshfeld表面分析,我们发现只有在分子间铜(I)–铜(I)距离约0.28 nm的Br–α中才存在Br―Br卤键及由其构成的卤素四聚体合成子,而其它两种晶体中仅存在Br–作用或弱氢键(如Br―H)。由此推测Br―Br卤键的存在可能是形成极短铜(I)–铜(I)距离的关键。
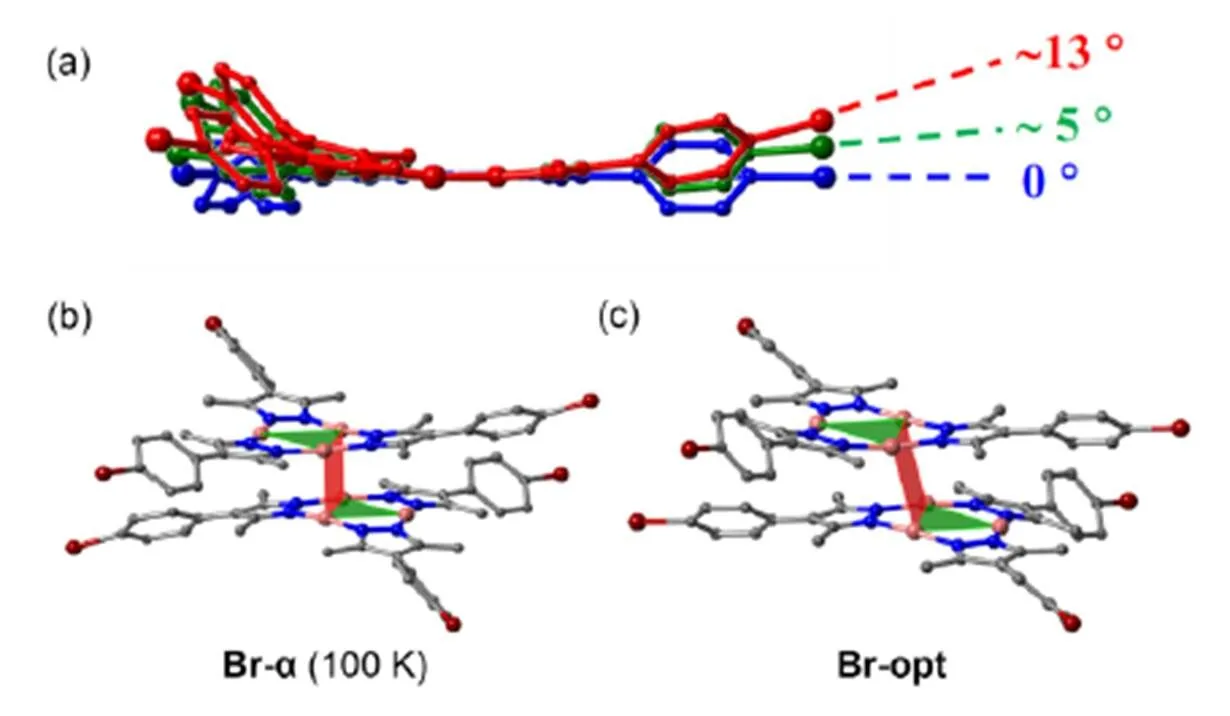
图4 (a)Br–α的分子弯曲,以红色、绿色和蓝色分别显示覆盖在一起的Br–α (100 K)、优化的二聚体及单体的模型;(b)Br–α (100 K)的晶体结构中的二聚体;(c) 优化的二聚体结构28
利用量子化学理论计算,选取所有晶体中提取出的二聚体模型及其优化构型进行比较,发现优化构型中分子间铜(I)–铜(I)距离接近0.310 nm,且二聚体模型的总能量与铜(I)–铜(I)距离之间并没有明显关系。这说明配体本身不会令配合物倾向于形成很短的铜(I)–铜(I)距离。
基于Mayer键级32、约化密度梯度(RDG)33以及分子中原子的量子理论(AIM)34–38,得知这些二聚体模型中非配体支撑的亲铜作用基本随着铜(I)–铜(I)距离的缩短而增强,且变化范围较大,最弱的铜(I)–铜(I)作用属于范德华力,而最强的已达到接近氢键的强度。相比之下,环三核分子内配体支撑的亲铜作用的强度基本不变,这在所有模型中都很相似,其强度也都接近范德华力的水平。AIM分析表明这些模型中非配体支撑的铜(I)–铜(I)作用总为典型的闭壳层作用。类似的,Dinda和Samuelson39对一系列单核亚铜配合物的二聚体模型进行AIM分析,也发现铜(I)–铜(I)作用总为闭壳层作用主导,只是一些模型中的铜(I)–铜(I)作用带有少许共价性质。这与当前一般认为的10金属离子的基态亲金属作用被色散力主导的结论一致。这样的作用力本质似乎与铜(I)–铜(I)距离关系不大,毕竟,我们的溴基配合物的分子间铜(I)–铜(I)距离可以很接近铜原子的范德华半径之和,而Samuelson等的配体支撑或非支撑的铜(I)–铜(I)距离甚至大部分都小于范德华半径之和,依然呈现出闭壳层作用主导的铜(I)–铜(I)作用本质。值得一提的是,我们的溴基模型中分子间铜(I)–铜(I)距离最远的情况下铜原子间相距0.3276 nm,此时不存在对应铜(I)–铜(I)作用的键临界点,RDG分析显示此时的分子间铜(I)–铜(I)作用为较弱的范德华力,因此对于上文提到的六核笼状化合物,不应期待其存在明显的环三核单元间铜(I)–铜(I)作用。
既然配体本身并不会在很大程度上导致配合物倾向于形成很强的亲铜作用,那么卤素原子在其中扮演了什么角色呢?为了探索其本质,我们针对卤键进行了计算。通过对构成卤素四聚体的四个配合物单体计算静电势,可以发现Br原子在背离C―Br键的区域存在一小块带正静电势的区域,而Br原子表面的其它区域都带负的静电势。此处的正静电势区域即卤键给体的穴,这就是Br―Br卤键具有很强方向性的本质原因。
图5列出了针对卤素四聚体的电子结构分析结果,其中AIM和RDG分析表明(图5(a, b)),Br―Br距离最短的卤键确实比距离较远的Br―Br作用更强,静电势分析(图5(c))发现每个Br原子的带正(负)静电势区域可以较好地与另一个Br原子的带负(正)静电势的区域匹配,而其它的Br―Br作用中Br原子之间的异号静电势区域不能很好地重叠,这也就解释了为何距离最短的卤键,其强度最大。对卤素四聚体进行AIM分析的结果也证明此处的Br―Br作用总为典型的闭壳层作用,且NPA电荷分析表明形成Br―Br作用不会明显改变Br原子的电子密度分布。
Br–α中极短的铜(I)–铜(I)距离的出现,一方面得益于Br―Br键的强方向性,另一方面也在于配体的刚性较强。配合物分子必须发生扭曲才能让每个分子提供两个溴原子参与形成卤素四聚体,而强刚性的配体却极难扭曲,因此只能令使配位键扭曲从而压迫环三核单元,令铜原子彼此靠近。假如吡唑环和苯环之间经由亚甲基或甚至更长的饱和烃基相连,则即使形成Br―Br卤键,对环三核单元的压迫也会弱得多,此时铜(I)–铜(I)距离也就不会如此的短。此例中卤键和亲铜作用属于两种彼此正交的作用力,类似的例子还有Panda等40最近报道的平面型六氯苯,其分子层面的卤―卤键与–作用令晶体可以在外界刺激下产生弯曲。作为一种本质与氢键类似的超分子作用,卤键在晶体工程、生物学等方面已经受到越来越多的关注41–43,而且已被发现可以有效的通过卤键调控发光量子产率和颜色44,45。
那么,如此短的铜(I)–铜(I)距离,是否会显著影响发光性质?我们对溴基配合物的一对超分子异构体进行常温固态发射光谱测试,尽管这两种晶体中的分子间铜(I)–铜(I)距离相差超过0.015 nm,但其发射光谱很大程度地重叠,最大发射峰都在670 nm左右,这样的发射能量,在已报道的环三核亚铜配合物的晶态磷光中偏低,但并没有显示出明显的差别。目前关于发光环三核亚铜配合物体系的研究,已发现低能磷光的出现需要足够短的分子间铜(I)–铜(I)距离,但发射波长与铜(I)–铜(I)距离之间的关系仍缺乏明确的结论,还需要更广泛的分子结构数据、深入的光谱研究和系统的理论计算积累。
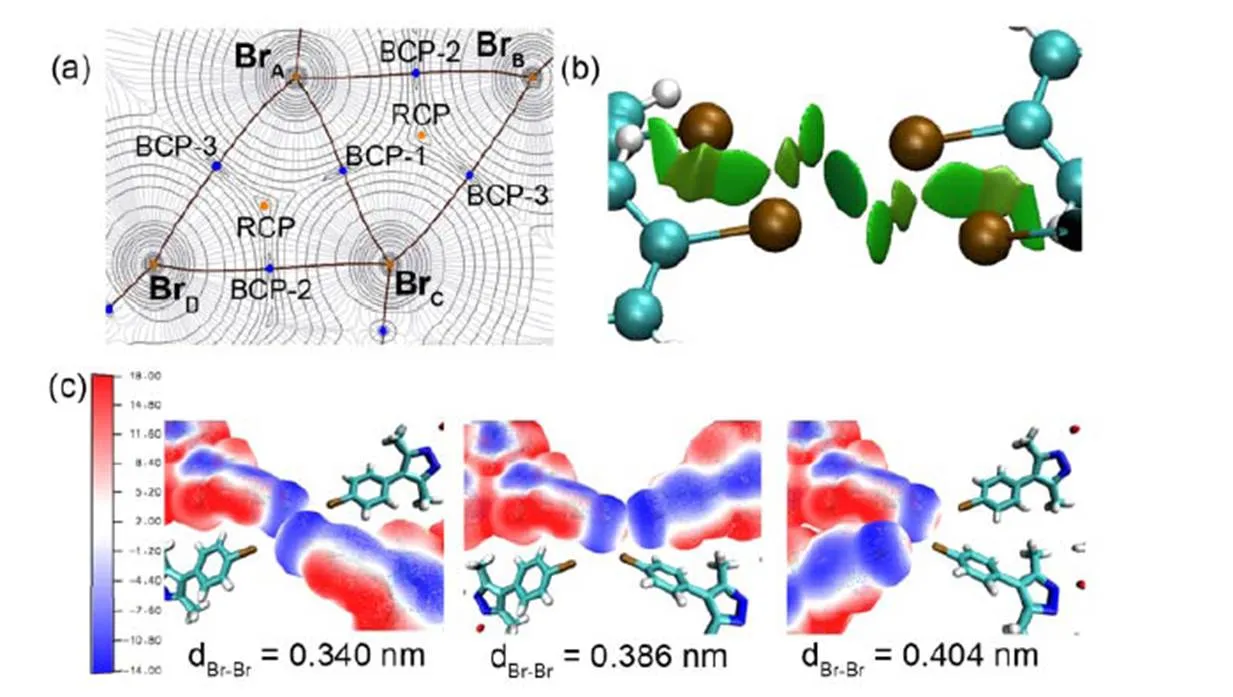
图5 Br–α(100 K)的Br四聚体的(a)AIM,(b)RDG和(c)静电势(以kcal∙mol−1为单位)分析的结果28
4 结论
亲金属作用在晶体工程及超分子构筑中日益受到关注,虽经历数十年研究,但仍存争论。本文结合本课题组近年来在吡唑基环三核亚铜配合物领域的部分研究成果,一方面通过有机连接体的作用得到具有环三核基元的六核笼状配合物,从而实现基态亲金属作用极弱但激发态亲金属作用非常显著的现象,其具有与离散型的环三核配合物不同的堆积模式及发光行为,并顺利将其中一种六核配合物与Cu2I2簇结合,设计为发光分子温度计。另一方面,也发现可以通过方向性强的Br―Br卤键及刚性较强的配体,实现此前环三核领域中前所未有的极短分子间铜(I)–铜(I)距离。然而,即使在铜(I)–铜(I)距离接近铜的范德华半径和的前提下,铜(I)–铜(I)作用依然表现为典型的闭壳层作用。这是通过共存的多种正交的超分子作用力构筑具有独特结构、性质的超分子实体或晶态材料的一个新的例子。
(1) Schmidbaur, H.; Schier, A.2008,, 1931. doi: 10.1039/B708845K
(2) Schmidbaur, H.; Schier, A.2012,, 370. doi: 10.1039/C1CS15182G
(3) Zhou, J.; Tan, X.; Hu, F.; Zou, H. H.; Paz, F. A. A.; Fu, L.; Zhao, R.. 2016,, 11292. doi: 10.1039/C6DT00883F
(4) Petty, J. T.; Sergev, O. O.; Ganguly, M.; Rankine, I. J.; Chevrier, D. M.; Zhang, P.2016,, 3469. doi: 10.1021/jacs.5b13124
(5) Su, C. Y., Pan, M., 1st ed.; Science Press: Beijing, 2010; pp 81–83. [苏成勇, 潘 梅. 配位超分子结构化学基础与进展. 北京: 科学出版社, 2010: 81–83.]
(6) Yadav, D.; Siwatch, R. K.; Mukherjee, G.; Rajaraman, G.; Nagendran, S.2014,10054. doi: 10.1021/ic5008389
(7) Yam, V. W. W.; Lo, Au, V. K. M.; Leung, S. Y. L20157589. doi: 10.1021/acs.chemrev.5b00074
(8) Che, C. M.; Lai, S. W.2005,, 1296. doi: 10.1016/j.ccr.2004.11.026
(9) Phillips, D. L.; Che, C. M.; Leung, K. H.; Mao, Z.; Tse, M. C.2005,, 1476. doi: 10.1016/j.ccr.2004.09.015
(10) Tsipis, A. C.2017,, 229. doi: 10.1016/j.ccr.2016.08.005
(11) Dias, H. V. R.; Polach, S. A.; Wang, Z... 2000,, 163. doi: 10.1016/S0022-1139(99)00313-9
(12) Dias, H. V. R.; Diyabalanage, H. V. K.; Eldabaja, M. G.; Elbjeirami, O.; Rawashdeh-Omary, M. A.; Omary, M. A.2005,, 7489. doi: 10.1021/ja0427146
(13) Vorontsov, I. I.; Kovalevsky, A. Yu.; Chen, Y. S.; Graber, T.; Gembicky, M.; Novozhilova, I. V.; Omary, M. A.; Coppens, P.2005,, 193003. doi: 10.1103/PhysRevLett.94.193003
(14) Hu, B.; Gahungu, G.; Zhang, J.2007,, 4965. doi: 10.1021/jp0689215
(15) Kim, K. H.; Kim, J. G.; Nozawa, S.; Sato, T.; Oang, K. Y.; Kim, T. W.; Ki, H.; Jo, J.; Park, S.; Song, C.; Sato, T.; Ogawa, K.; Togashi, T.; Tono, K.; Yabashi, M.; Ishikawa, T.; Kim, J.; Ryoo, R.; Kim, J.; Ihee, H.; Adachi. S.2015,, 385. doi: 10.1038/nature14163
(16) Jarzembska, K. N.; Kaminski, R.; Fournier, B.; Trzop, E.; Sokolow, J. D.; Henning, R.; Chen, Y.; Coppens, P.2014,, 10594. doi: 10.1021/ic501696y
(17) Grimes, T.; Omary, M. A.; Dias, H. V. R.; Cundari, T. R.2006,, 5823. doi: 10.1021/jp0605146
(18) Nitsch, J.; Lacemon, F.; Lorbach, A.; Eichhorn, A.; Cisnetti, F.; Steffen, A.2016,, 2932. doi: 10.1039/C5CC09659F
(19) He, J.; Yin, Y. G.; Wu, T.; Li, D.; Huang, X. C.2006,, 2845. doi: 10.1039/B601009A
(20) Xiao, Q.; Zheng, J.; Li, M.; Zhan, S. Z.; Wang, J. H.; Li, D.2014,, 11604. doi: 10.1021/ic5016687
(21) Ni, W. X.; Li, M.; Zheng, J.; Zhan, S. Z.; Qiu, Y. M.; Ng, S. W.; Li, D.2013,, 13472. doi: 10.1002/anie.201308135
(22) Ni, W. X.; Qiu, Y. M.; Li, M.; Zheng, J.; Sun, R. W. Y.; Zhan, S. Z.; Ng, S. W.; Li, D.2014,, 9532. doi: 10.1021/ja5025113
(23) Gao, G. F.; Li, M.; Zhan, S. Z.; Lv, Z.; Chen G. H.; Li, D.2011,, 4113. doi: 10.1002/chem.201100081
(24) Wang, J. H.; Li, M.; Zheng, J.; Huang, X. C.; Li, D.2014,, 9115. doi: 10.1039/C4CC04100C
(25) Zhan, S. Z.; Li, M.; Zhou, X. P.; Li, D.; Ng, S. W.2011,, 1457. doi: 10.1039/C1RA00566A
(26) Zhan, S. Z.; Li, M.; Zhou, X. P.; Wang, J. H.; Yang, J. R.; Li, D.2011,, 12441. doi: 10.1039/C1CC14303D
(27) Zhan, S. Z.; Li, M.; Ng, S. W.; Li, D.2013,, 10217. doi: 10.1002/chem.201204632
(28) Wang, X. L.; Zheng, J.; Li, M.; Ng, S. W.; Chan, S. L. F.; Li, D.2016,, 4991. doi: 10.1021/acs.cgd.6b00571
(29) Tong, Z. H., Zhang, J. C., Zhu, J. C., Fan, M. G., Fu, H. B., Wu, H. B., Wu, Y. S., Xu, J. H., Zhu, A. P., Fan, P., Fu, W. F., Chi, S. M., Liu, C. Y., 1st ed.; Science Press: Beijing, 2013; pp 533–539. [佟振合, 张建成, 朱晋昌, 樊美公, 付红兵, 吴义室, 徐建华, 朱爱平, 樊平, 傅文甫, 迟绍明, 刘春艳. 分子光化学. 北京: 科学出版社, 2013: 533–539.]
(30) Leitl, M. J.; Kuchle, F. R.; Mayer, H. A.; Wesemann, L.; Yersin, H.2013,, 11823. doi: 10.1021/jp402975d
(31) Singh, K.; Long, J. R.; Stavropoulos, P.1997,, 2942. doi: 10.1021/ja963664a
(32) Mayer, I.1983,, 270. doi: 10.1016/0009-2614(83)80005-0
(33) Johnson, E. R.; Keinan, S.; MoriSánchez, P.; ContrerasGarcía, J.; Cohen, A. J.; Yang, W.2010,, 6498. doi: 10.1021/ja100936w
(34) Bader, R. F. W.2007,, 7966. doi: 10.1021/jp073213k
(35) Bader, R. F. W.1991,, 893. doi: 10.1021/cr00005a013
(36) Jenkins, V.; Morrison, I.2000,, 97. doi: 10.1016/S0009-2614(99)01306-8
(37) Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E.2002,, 5529. doi: 10.1063/1.1501133
(38) Varadwaj, P. R.; Marques, H. M.2010,, 2126. doi: 10.1039/B919953E
(39) Dinda, S.; Samuelson, A. G.2012,, 3032. doi: 10.1002/chem.201101219
(40) Panda, M. K.; Ghosh, S.; Yasuda, N.; Moriwaki, T.; Mukherjee, G. D.; Reddy, C. M.; Naumov, P.2015,, 65. doi: 10.1038/nchem.2123
(41) Persch, E.; Dumele, O; Diederich, F.2015,, 3290. doi: 10.1002/anie.201408487
(42) Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G.2016,, 2478. doi: 10.1021/acs.chemrev.5b00484
(43) Liu, M. H.2017,, 451. [刘鸣华. 物理化学学报, 2017,, 451.] doi: 10.3866/PKU.WHXB201702151
(44) Sivchik, V. V.; Solomatina, A. I.; Chen, Y. T.; Karttunen, A. J.; Tunik, S. P.; Chou, P. T.; Koshevoy, I. O.2015,, 14057. doi: 10.1002/anie.201507229
(45) Yan, D.; Delori, A.; Lloyd, G. O.; Friščić, T.; Day, G. M.; Jones, W.; Lu, J.; Wei, M.; Evans, D. G.; Duan, X.2015,, 12483. doi: 10.1002/anie.201106391
Extreme Long and Extreme Short Cu–Cu Distances: Nature of Cuprophilicity and Luminescence Modification in Cu(I)-based CyclicTrinuclear Complexes
ZHENG Ji1XING Li-Rui2LI Dan1,*
(1,,;2,,)
This monograph focuses on recent progress in the research field of metallophilicity, in combination of our latest results on Cu(I)-based cyclic trinuclear complexes, mainly discussing two examples with very different cuprophilicity. One is by constructing triangular coordination prisms bearing cyclic trinuclear units, confirming that strong cuprophilic attraction could exist in the phosphorescent emissive state under frontal packing mode even when the cuprophilicity is extreme weak at ground state, and by coordination with a Cu2I2cluster with an additional coordinate site in the ligand, so that a self-calibrated wide-range luminescent molecular thermometer was obtained. The other is the realization of the shortest Cu–Cu distance so far in this field by Br–Br halogen bond orthogonal to cuprophilicity, followed by the investigation using several kinds of electronic structure analysis. The result shows that even when the Cu–Cu distance approaches the van der Waals radii sum of Cu, its nature is still closed-shell interaction, which is also the nature of all Br–Br interaction, and the strongest Br–Br interaction in this system displays good matching between the-hole of a Br atom and the belt of negative potential of another Br atom.
Cuprophilicity; Frontal packing; Phosphorescent emissive state; Halogen bond;Reduced density gradient; Quantum theory of atoms in molecules
April 26, 2017;
May 26, 2017;
June 5, 2017.
Corresponding author. Email: danli@jnu.edu.cn; Tel: +86-20-85221697.
10.3866/PKU.WHXB201706051
O641
The project was supported by the National Key Basic Research Program of China (973) (2012CB821706, 2013CB834803), the National Natural Science Foundation of China (91222202, 21171114)
国家重点基础研究发展计划(973) (2012CB821706, 2013CB834803)和国家自然科学基金(91222202, 21171114)资助项目
猜你喜欢
数学物理学报(2022年3期)2022-05-25
数学物理学报(2022年1期)2022-03-16
今日农业(2021年2期)2021-11-27
数学物理学报(2021年5期)2021-11-19
数学物理学报(2021年3期)2021-07-19
内燃机工程(2021年1期)2021-02-05
今日农业(2020年23期)2020-12-31
含能材料(2020年7期)2020-07-11
分析化学(2017年12期)2017-12-25
分析化学(2017年12期)2017-12-25
