
高效双功能Au@SH/SO3H-MSiO2-S催化剂的制备及其性能研究
2018-01-12 09:56:26嵇小荷方海洁吴佳敏孙小军
淮阴师范学院学报(自然科学版) 2017年4期
嵇小荷, 方海洁, 邓 永, 吴佳敏, 孙小军
(淮阴师范学院 化学化工学院, 江苏 淮安 223300)
0 引言
硅醇是含有Si-OH的化合物,其在自然界中普遍存在.在有机合成中具有重要的作用,其可以用作硅基聚合物材料的基石[1-2]以及作为有机合成反应中的亲核耦合剂[3-5],因此被广泛应用于有机转化反应中.尽管已报道了许多硅醇的制备方法,但是比较理想的方法是用负载型纳米金属非均相催化剂催化有机硅烷与水的氧化反应.因为从环境友好角度考虑,这种类型的催化剂可以重复循环使用,且生成的另外一种产物是氢气.如Kaneda等报道羟基磷灰石负载的金纳米粒子是硅烷氧化反应的高效催化剂.但是固载化的金纳米粒子在催化硅烷氧化反应过程中容易流失[6].通常采用焙烧法提高载体和金属之间相互作用解决金纳米粒子流失的方法,然而,在高温条件下进行焙烧容易导致较小的金纳米粒子团聚,使其催化活性降低.至今为止,如何稳定较小的金纳米粒子仍然面临着挑战.
本文采用共聚法制得巯基功能化的短孔道有序介孔材料SH-MSiO2-S,通过介孔材料孔道中的巯基原位还原HAuCl4中的Au(III),分别生成磺酸基(-SO3H)纳米金粒子,该金纳米粒子与未发生反应的巯基(-SH)络合从而固定在载体上,从而得到Au@SH/SO3H-MSiO2-S催化剂.该催化剂在水介质中三乙基硅烷水解反应中,表现出较好的催化性能.
1 实验部分
1.1 试剂与仪器
聚环氧乙烯醚-聚环氧丙烯醚-聚环氧乙烯醚(P123,EO20PO70EO20,Mw=5 800)及3-巯丙基三乙氧基硅烷(SHCH2CH2CH2Si(OC2H5))购自Sigma-Aldrich公司;三乙基硅烷、正硅酸乙酯(Si(OC4H9)4)、三乙基硅烷((C2H5)SiH)、氯氧化锆(ZrOCl2)及丙酮等购自国药集团化学试剂有限公司.样品晶相结构由XRD(Rigaku D/max-RB, Cu Kα)确定,样品的孔道结构和表面形貌由透射电子显微镜(TEM,JEOL JEM2011);通过场发射扫描电子显微镜(FESEM,Hitachi S4800)观察样品的表面形貌.通过Quantachrome公司生产的NOVA 4000e分析仪测得的N2吸附-脱附等温线,根据BET和BJH模型计算得到样品的结构参数.采用ICP(Varian VISTA-MPX)分析确定样品中Au的负载量(wt%).
1.2 催化剂的制备
1.2.1 短孔道巯基功能化介孔材料的制备(SH-MSiO2-S)
在100 mL圆底烧瓶中依次加入40 mL 2.0 mol/L HCl溶液、1.0 g P123和0.16 g ZrOCl2·8H2O,并将其置于35℃水浴中搅拌.待P123充分溶解后,将一定量的TEOS逐滴加入上述溶液中,然后在35℃条件下使TEOS水解后继续搅拌90 min后,向其中加入一定量的3-巯丙基三乙氧基硅烷,控制反应体系中P123与总硅量(TEOS与3-巯丙基三乙氧基硅烷中的硅)的摩尔比0.017:10,然后在35℃的条件下继续搅拌24 h,再将此混合物放进水热釜中,于100℃陈化24 h.经抽滤、洗涤、干燥后,用酸化的乙醇溶液在80℃除去表面活性剂.即得短孔道巯基功能化介孔材料SH-MSiO2-S.
1.2.2 Au@SH/SO3H-MSiO2-S催化剂的合成
将1.0 g SH-MSiO2-S短孔道介孔材料加入到一定量的无水乙醇和氯金酸水溶液中,然后在25℃下搅拌24 h,溶液由淡黄色逐渐变成无色.最后经过滤、大量乙醇洗涤多次、真空干燥即得催化剂Au@SH/SO3H-MSiO2-S.样品中Au的含量采用电感耦合等离子体光谱(ICP)测量.
1.3 催化剂活性测试

反应式1三乙基硅烷氧化反应
选用水相三乙基硅烷氧化反应(反应式1)为探针,考察Au@SH/SO3H-MSiO2-S催化剂的催化性能.具体反应过程如下:向25 mL反应瓶中加入0.5 mmol三乙氧基硅烷、、正辛烷(内标物)、少量Au@SH/SO3H-MSiO2-S催化剂、2.0 mL H2O,在25℃下搅拌反应.反应结束后,向反应瓶中加入一定量的乙酸乙酯萃取出反应体系中的有机物.萃取出的有机物用气相色谱仪GC(日本岛津GC2014)进行定量分析,色谱检测条件为:色谱柱采用非极性的JWDB-5(L=15 m,∅=0.25 mm),,以N2为载气,氢火焰为检测器,柱温由80℃升到250℃,升温速率为10℃/min.采用内标法确定反应的转化率和得率.实验中所有催化活性数据都重复3次以上,数据的误差范围在±5%以内.
2 结果与讨论
2.1 催化剂的表征
图1为不同催化剂的FESEM图,从图中可以看出,催化剂Au@SH/SO3H-MSiO2-S呈规整的棒状,棒的长度约为400 nm,其介孔孔道清晰可见,孔道的长度为300~400 nm(见图1).从图2的样品HRTEM图可见,所有样品都具有规整的有序介孔结构,样品的小角XRD图(图3)和N2吸附-脱附等温线(图4)上得到进一步证明了样品介孔结构的存在.

图1 Au@SH/SO3H-MSiO2-S样品的FESEM图
从图3可以看出,不同负载量的催化剂在2θ=0.7~0.9o均出现1个较强的小角衍射峰对应于样品的[100]面,在2θ=1.2~2.0o出现了2个弱衍射峰,分别对应于样品的[110]和[200]面,这些结果表明样品具有二维六方介孔结构[7].从Au@SH/SO3H-MSiO2-S-1到Au@SH/SO3H-MSiO2-S-9,随着样品中Au含量的增加,小角XRD中[100]面的衍射峰强度有所减弱,高角度出现的2个弱衍射峰随着负载量的增加逐渐消失,这表明巯基硅烷会影响表面活性剂的自组装过程,不利于样品介孔结构的形成,从而使样品的有序性降低[8].

图2 Au@SH/SO3H-MSiO2-S样品的HRTEM图
图4给出了Au@SH/SO3H-MSiO2-S-样品的N2吸附-脱附等温线图.从图4可见,样品表现出典型的第IV类型N2吸附-脱附等温线,有H1型的滞后环,在p/p0=0.4~0.55处有1个比较明显的毛细凝聚突跃,这进一步证明了此样品具有有序介孔结构[9].

图3 不同样品的小角XRD图 图4 Au@SH/SO3H-MSiO2-S-9样品的N2吸附-脱附等温线图
图5是Au@SH/SO3H-MSiO2-S的XPS图谱.从S2p的图中可以看出,SH-MSiO2-S与HAuCl4相互作用之后,所得到的Au@SH/SO3H-MSiO2-S催化剂在164.2 eV和169.8 eV处出现了2个峰,分别归属于S的-2价态及+6价态即对应于催化剂中的巯基和磺酸.Au@SH/SO3H-MSiO2-S中金的Au4f在84.6 eV和88.3 eV处出峰,两者的Au4f的结合能之差为3.70 eV,由此可以看出催化剂中金是以金属态金的形式存在,这也进一步证实了高分散的金纳米粒子催化剂被成功制备.
2.2 催化剂的性能测试
反应结束后通过GC-MAS检测反应体系,结果表明,催化剂对目标产物的三乙氧基硅醇的选择性为100%.从表1中不同催化剂的活性可以看出,载体HS-MSiO2-S中在反应中呈现惰性,而催化剂Au@SH/SO3H-MSiO2-S没有添加剂存在的条件下,能顺利地催化水介质中三乙基硅烷氧化成三乙基硅醇,这可归因于Au@SH/SO3H-MSiO2-S催化剂中的Au纳米粒子高度分散,具有较高的反应活性.有趣的是,若使用Au/SH-MSiO2-S催化剂,此反应不能发生;但加入少量的H2SO4到此反应体系中,反应可以进行,但在25℃反应30 min后,三乙基硅烷的转化率只有15.5%.由此可见,硅烷氧化反应不仅需要金催化活性中心,还要酸作为共催化剂.由此可以看出,Au@SH/SO3H-MSiO2-S非均相催化剂是一个双功能催化剂,即具有反应需要的金属和酸催化活性中心.
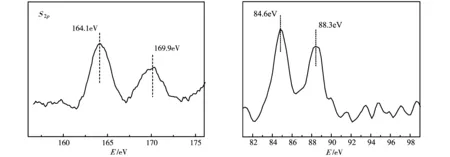
图5 Au@SH/SO3H-MSiO2-S样品中S的XPS图
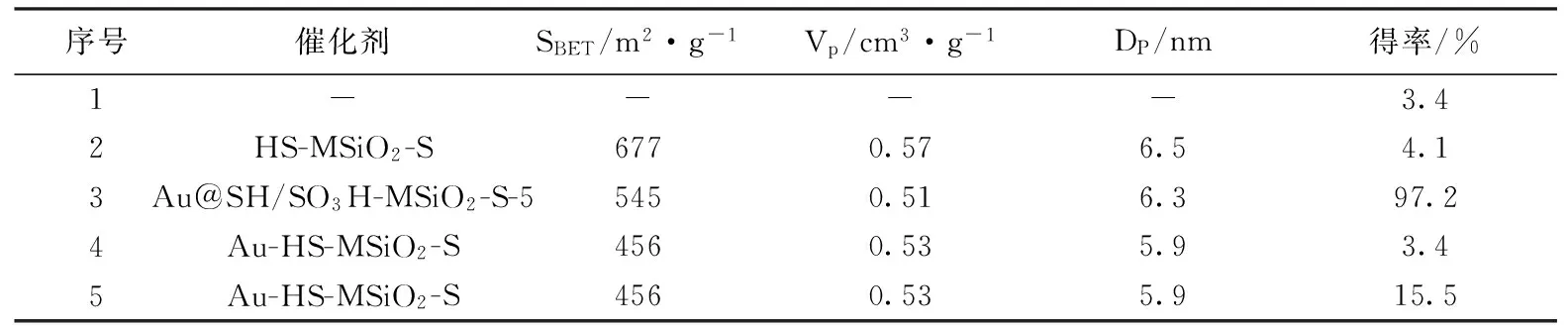
序号催化剂SBET/m2·g-1Vp/cm3·g-1DP/nm得率/%1----3.42HS-MSiO2-S6770.576.54.13Au@SH/SO3H-MSiO2-S-55450.516.397.24Au-HS-MSiO2-S4560.535.93.45Au-HS-MSiO2-S4560.535.915.5
反应条件:1.0 mmol硅烷,1.5 mL溶剂,25℃,30 min;0.50 mmol H2SO4.

表2 金负载量对催化剂活性的影响
反应条件:1.0mmol硅烷,0.10mLH2O,1.5mLH2O,30min.
表2是金负载量对催化剂活性的影响.表中Au@SH/SO3H-MSiO2-S-1表示以1%SH-MSiO2-S为载体制得的催化剂,依次类推.随着载体中巯基含量的增加,其通过与氯金酸发生原位氧化还原负载到载体中金含量随之增加.从表中可以看出,当载体中巯基量较少时,催化剂的活性较低,得率仅49.0%,随着载体中巯基的含量增加,产物得率随之增加,当使用SH-MSiO2-S-5为载体制得的催化剂产物得率达97.2%,如金的负载量进一步增加,产物得率没有明显提高,所以确定Au@SH/SO3H-MSiO2-S-5为最佳催化剂.
表3给出了催化剂用量和温度反应条件对硅烷氧化反应活性的影响.从表中看出,随着催化剂用量的增加目标产物的得率也随之逐渐增加,当催化剂用量增加至0.10g,产物的转化率达到最佳,99.9%;而反应温度三乙基硅醇的得率影响比较明显,25℃为最佳反应温度,超过25℃,随着温度上升反应得率下降.

表3 反应条件对Au@SH/SO3H-MSiO2-S-5催化剂活性的影响
反应条件:1.0 mmol硅烷,0.10 mL H2O,1.5 mL H2O,30 min.
3 结论
成功制备了高效短孔道有序介孔负载型高分散的金纳米粒子催化剂Au@SH/SO3H-MSiO2-S,催化剂具有较好的热稳定性,在反应过程中不易团聚,对空气比较稳定,易从反应体系中分离.该催化剂在水介质三乙基硅烷的水解反应中显示了良好的催化性能,具有较好的应用前景.
[1] Chandrasekhar V, Boomishankar R, Nagendran S. Recent developments in the synthesis and structure of organosilanols[J]. Chem Rev, 2004, 104: 5847-5910.
[2] Murugavel R, Walawalkar M G, Dan M, et al. Transformations of molecules and secondary building units to materials:A bottom-up approach[J]. Acc Chem Res, 2004, 37:763-774.
[3] Hirabayashi K, Nishihara Y, Mori A, et al. A novel C-C bond forming reaction of aryl-and alkenylsilanols. A halogen-free Mizoroki-Heck type reaction[J].Tetrahedron Lett, 1998, 39: 7893-7896.
[4] Hirabayashi K, Kawashima J, Nishihara Y, et al. A new transformation of silanols. Palladium-catalyzed cross-coupling with organic halides in the presence of silver(I) oxide[J]. Org Lett, 1999, 1: 299-301.
[5] Denmark S E, Wehrli D. Highly stereospecific, palladium-catalyzed cross-coupling of alkenylsilanols[J]. Org Lett, 2000, 2: 565-568.
[6] Kang T, Park Y, Yi J. Highly selective adsorption of Pt2+and Pd2+using thiol-functionalized mesoporous silica[J]. Ind Eng Chem Res, 2004, 43: 1478-1484.
[7] Zhao D Y, Huo Q S, Feng J L, et al. Nonionic triblock and star diblock copolymer and oligomeric surfactant syntheses of highly ordered, hydrothermally stable, mesoporous silica structures[J]. J Am Chem Soc,1998, 120: 6024-6036.
[8] Wahab M A, Ha C S. Hybrid Periodic mesoporous organosilica materials prepared from 1,2-bis(triethoxysilyl)ethane and (3-cyanopropyl)triethoxysilane[J]. Micropor Mesopor Mater, 2004, 69: 19-27.
[9] Hu Q Y, Hampsey J E, Jiang N, et al. Surfactant-templated organic functionalized mesoporous dilica with phosphino ligands[J]. Chem Mater, 2005, 17: 1561-1569.
[10] Ilieva L, Sobczak J W, Manzoli M, et al. Reduction behavior of nanostructured gold catalysts supported on mesoporous titania and zirconia. Appl Catal A: Gen, 2005, 291: 85-92.
[11] Han J, Liu Y, Guo R. Facile synthesis of highly stable gold nanoparticles and their unexpected excellent catalytic activity for Suzuki-Miyaura cross-coupling reaction in water[J]. J Am Chem Soc, 2009, 131: 2060-2061.
猜你喜欢
化学工程师(2023年1期)2023-02-17 15:09:48
理化检验-化学分册(2020年12期)2021-01-26 00:41:38
中国果树(2020年2期)2020-07-25 02:14:28
上海农业科技(2019年1期)2019-02-22 01:51:28
中国资源综合利用(2016年7期)2016-02-03 03:00:19
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01 02:54:14
四川师范大学学报(自然科学版)(2015年1期)2015-02-28 14:07:29
海洋科学进展(2015年1期)2015-02-27 13:16:16
火炸药学报(2014年5期)2014-03-20 13:17:51
食品科学(2013年23期)2013-03-11 18:30:02
