
离线二维液相色谱法分离巴天酸模根的化学成分
2018-01-11 07:40赵雅清付冬梅刘艳芳梁鑫淼薛慧清
色谱 2018年1期
赵雅清, 付冬梅, 刘艳芳, 梁鑫淼, 薛慧清*
(1. 山西中医药大学, 基于炎性反应的重大疾病创新药物山西省重点实验室, 山西 太原 030001; 2. 中国科学院大连化学物理研究所, 中国科学院分离分析化学重点实验室, 辽宁 大连 116021)
中医药学凝聚了中华民族几千年的健康养生理念与实践经验,是中国古代科学的珍宝,深入研究中医药学对于推动生命科学以及医药事业的发展意义深远。然而,由于中药成分的复杂性,从常量成分(mg/g)到微量组分(pg/g)通常包含上百种化合物,因此对中药组分的分离、分析是当前中药分析领域的重点和难点。对于中药复杂样品的分离,传统的一维色谱法往往不能提供足够的分辨率和峰容量,色谱峰重叠现象严重;而通过串联两种或两种以上不同分离机制的色谱柱,构建多维色谱分离系统,则可以极大地提高对复杂中药样品的分离能力[1]。
与一维液相色谱法相比,二维液相色谱法(2D- LC)具有更高的峰容量以及分离度,对复杂体系以及低含量物质的分离有明显的优势[2]。二维色谱有离线和在线两种方式。与离线模式相比,在线模式在技术上仍存在一定的挑战,如流动相的兼容性、分离速度的兼容性、一维与二维洗脱强度的兼容性等问题[3,4]。离线二维色谱法适用成本低,溶剂选择性广,峰容量大[5],每一维的分离条件可独立优化,同时第二维没有分析时间的限制,在优化的条件下能够达到最佳的分离能力,且不受流动相兼容问题的影响[6]。
目前,随着色谱柱填料类型的多样化、商品化,以及高压/超高压液相色谱系统的出现和数据软件的完善,建立二维液相色谱方法更加简便,应用范围更广[7]。基于制备型液相色谱的中药化学组分制备技术已广泛应用于中药的物质基础研究[8]。Jin等[9]利用C3和C18色谱柱构建2D- LC系统,对忍冬的各成分进行了分离制备,结果获得了纯度大于99%的7个化合物。Fan等[10]建立了2D- 正相液相色谱/反相液相色谱方法分离制备甘草中的黄酮类化合物,共得到24个高纯度化合物。贾有梅等[11]建立了亲水/反向二维液相制备色谱分离纯化络石藤中化学成分的方法,最终得到14个高纯度化合物。Li等[12]应用离线二维液相色谱对荜拨中的化学成分进行分离,最终得到25个生物碱化合物。Guo等[13]使用XUnion C18与XAmide色谱柱构建离线二维色谱,对三七叶子的25个一维目标流分进行分离,最终得到8个皂苷成分。二维液相色谱对样品有预处理、分离的功能,能在短时间内实现高峰容量,越来越多地运用于中药成分的分离分析[14]。鉴于离线二维色谱不涉及二维色谱之间的阀切换,对仪器要求不高,对复杂样品具有较好的分离能力和分辨率,本文选用巴天酸模作为模型中药建立离线二维液相色谱分离体系。
巴天酸模(RumexpatientiaL.),蓼科酸模属植物,中药土大黄的植物来源之一,具有化痰止咳、清热通便、凉血止血、燥湿杀虫止痒等功效[15],富含蒽醌、单宁、萘及萘醌衍生物[16]。刘景等[17]应用硅胶柱色谱、Sephadex LH- 20分离纯化巴天酸模根的化学成分,得到大黄酚- 8-O-β- D- 葡萄糖苷、大黄素- 6-O-β- D- 葡萄糖苷、大黄素甲醚- 8-O-β- D- 葡萄糖苷等7个单体化合物。迄今为止,对巴天酸模的分离仍采用硅胶柱层析等传统方法。
本文建立了反相/反相离线二维色谱分离系统,通过优化色谱柱类型和流动相条件,对巴天酸模根提取液乙酸乙酯萃取层一维分离的18个流分分别进行进一步的二维分析。本方法的建立为快速、有效地从巴天酸模根中制备标准品、筛选活性组分以及对潜在微量活性化合物的制备提供了技术支持。
1 实验部分
1.1 仪器、试剂与材料
Waters 2695高效液相色谱系统,包括四元梯度泵、自动进样器、柱恒温系统、2998光电二极管阵列检测器、2489双波长检测器和Empower工作站(美国Waters公司);环氧四氮唑色谱柱(250 mm×4.6 mm, 7 μm,批号:R2016053003,实验室自制); Unitary C18色谱柱(250 mm×4.6 mm, 7 μm,批号:Z2013040901,华谱新创科技有限公司);甲酸(色谱纯)购自美国Acros公司;甲醇(色谱纯)、石油醚(分析纯)和乙酸乙酯(分析纯)购自天津科密欧公司;超纯水由Milli- Q水纯化系统(美国Millipore公司)制备得到。
药材巴天酸模于2015年6月18日采自北京市延庆区井庄乡冯家庙村南通往莲花滩村的马路两侧,经中国医学科学院药用植物研究所郭宝林研究员鉴定为蓼科植物土大黄(巴天酸模属)。
1.2 样品的前处理
取巴天酸模根部药材粉碎,精密称取药材粉末10 g置于烧瓶中,依次用100、80、60 mL的95%乙醇分别超声提取,每次1 h,合并滤液(记为BTS- 醇),减压浓缩至不含乙醇,溶于150 mL水,依次用150 mL的石油醚、乙酸乙酯萃取,得到石油醚层(标记为BTS- PE)、乙酸乙酯层(标记为BTS- ET),萃取完剩余水层标记为BTS- 水。将各萃取液减压浓缩、冷冻干燥后,称取5~8 mg样品,加甲醇溶解,过0.45 μm微孔滤膜,作为分析样品。取乙酸乙酯萃取得到的分析样品,配制质量浓度为103.38 g/L的溶液,过0.45 μm微孔滤膜,作为一维分离的样品。
1.3 巴天酸模根提取液各萃取层的HPLC分析
用Waters 2695- 2998色谱系统分别对BTS- 醇、BTS- PE、BTS- ET和BTS- 水4个样品进行分析,采用Unitary C18柱(250 mm×4.6 mm)作为分析柱,流动相由0.1%(v/v)甲酸水溶液(A)和甲醇(B)组成。梯度洗脱条件:0~5 min, 5%B; 5~45 min, 5%B~95%B; 45~50 min, 95%B。流速:1.0 mL/min,柱温:30 ℃,检测波长:210~400 nm,进样量:10 μL。
1.4 二维分离条件
1.4.1一维色谱
用Waters 2695- 2489色谱系统对巴天酸模根乙酸乙酯层进行分离,采用环氧四氮唑色谱柱(250 mm×4.6 mm),流动相为0.1%(v/v)甲酸水溶液(A)和甲醇(B)。梯度洗脱条件:0~5 min, 25%B; 5~50 min, 25%B~90%B; 50~55 min, 90%B。流速:1.0 mL/min;柱温:30 ℃;检测波长:266 nm;上样量:30 μL。按峰收集流分F1~F18,共制备3次,合并相同流分,分别浓缩至约1 mL。
1.4.2二维色谱
用Waters 2695- 2998色谱系统对一维流分F1~F18进行分析,采用Unitary C18色谱柱(250 mm×4.6 mm),流动相为0.1%(v/v)甲酸水溶液(A)和甲醇(B);梯度洗脱条件:0~5 min, 25%B; 5~50 min, 25%B~90%B; 50~55 min, 90%B。流速:1.0 mL/min;柱温:30 ℃;检测波长:210~400 nm;上样量:20 μL。
2 结果与讨论
2.1 巴天酸模根各萃取层的HPLC分析
对药材的4个萃取层BTS- 醇、BTS- PE、BTS- ET和BTS- 水按照1.3节条件分别进行HPLC分析,结果见图1。
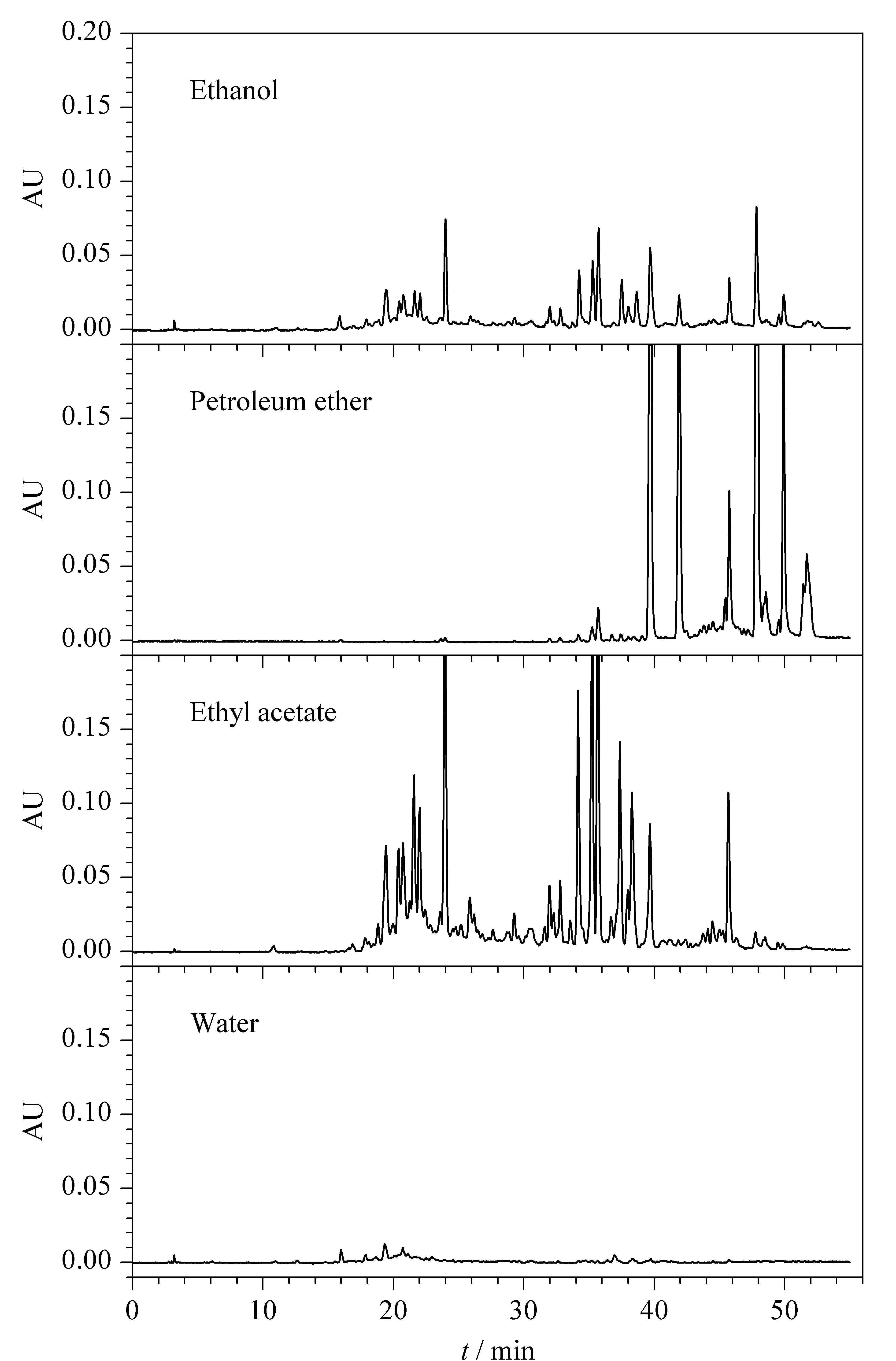
图 1 巴天酸模根提取液各萃取层样品的色谱图Fig. 1 Chromatograms of extracts of root of Rumex patientia L. sample with different solvents Column: Unitary C18 (250 mm×4.6 mm, 7 μm). Mobile phases: (A) 0.1% (v/v) formic acid aqueous solution and (B) methanol. Gradient elution: 0-5 min, 5%B; 5-45 min, 5%B-95%B; 45-50 min, 95%B. Detection wavelength: 210-400 nm. Injection volume: 10 μL. Flow rate: 1. 0 mL/min.
从图1中可以看出,巴天酸模根乙醇提取液成分复杂,涵盖药材中中等极性及弱极性成分;通过石油醚萃取,巴天酸模根提取液中大部分弱极性成分被分离出;乙酸乙酯层保留了巴天酸模根提取液大部分的中等极性成分;水层中可能保留一些没有紫外吸收的单糖和寡糖类成分,需要通过其他途径进一步分析。鉴于乙酸乙酯层中涵盖了乙醇提取液的大部分组分,且萃取的目标物含量高,因此选择对乙酸乙酯层进行第二维色谱分析。
2.2 色谱柱的选择及其保留特性的比较
在前期研究中,制备了一种苯基与四唑基键合在二氧化硅上的环氧四氮唑固定相材料,与常规C18材料具有不同的选择性与保留特性。在此基础上,本研究分别选用环氧四氮唑与Unitary C18材料装填的色谱柱,对巴天酸模根的乙酸乙酯提取液进行分析,结果见图2。

图 2 巴天酸模根乙酸乙酯萃取液在(a)环氧四氮唑和(b)Unitary C18色谱柱上的色谱图Fig. 2 Chromatograms of ethyl acetate extract of root of Rumex patientia L. on (a) phenyl/tetrazolium column and (b) Unitary C18 column a. Column: phenyl/tetrazolium (250 mm×4.6 mm, 7 μm). Mobile phases: (A) 0.1% (v/v) formic acid aqueous solution and (B) methanol. Gradient elution: 0-5 min, 10%B-25%B; 5-45 min, 25%B-90%B; 45-55 min, 90%B. Detection wavelength: 266 nm. Injection volume: 10 μL. Flow rate: 1. 0 mL/min. b. Column: Unitary C18 (250 mm×4.6 mm, 7 μm). Mobile phases: (A) 0.1% (v/v) formic acid aqueous solution and (B) methanol. Gradient elution: 0-5 min, 15%B; 5-50 min, 15%B-75%B; 50-55 min, 75%B. Detection wavelength: 266 nm. Injection volume: 10 μL. Flow rate: 1. 0 mL/min.
在图2a中环氧四氮唑色谱柱上2、3号色谱峰未分离,在图2b中Unitary C18色谱柱上基线分离。在环氧四氮唑色谱柱上分离得到的6~15号峰的出峰顺序在Unitary色谱柱上均有所改变,说明环氧四氮唑和Unitary C18这两种材料对样品的选择性不同,可以互补来构建分离效果较好的二维色谱系统。本文中利用环氧四氮唑色谱填料和C18键合色谱填料制作的不同色谱柱来构建离线二维色谱分离体系。为了方便后续的制备,采用了甲醇和水(含0.1%(v/v)甲酸)作为流动相优化分离条件,流动相体系简单,没有盐的引入,避免了脱盐处理。
图2a色谱图峰形简单,峰的重叠现象少,在上样量较大的一维制备中峰之间重合的可能性低,因此,选用环氧四氮唑作为一维色谱分离材料。第二维色谱分析需要色谱柱具有更好的分离效果,而图2b色谱图峰间距小,峰多且杂,选择Unitary C18作为二维色谱的分析柱,能够得到更多的样品信息。
2.3 巴天酸模根乙酸乙酯萃取液的一维色谱分离
在构建反相/反相二维色谱系统中,选择环氧四氮唑作为一维分离色谱柱。在上样量大的情况下,一些含量低的成分峰在制备谱图中比较明显,因此,实际接取流分的数量与分析谱图中峰的数量存在差异。按色谱峰接取流分,得到18个流分,分别为F1~F18(见图3)。
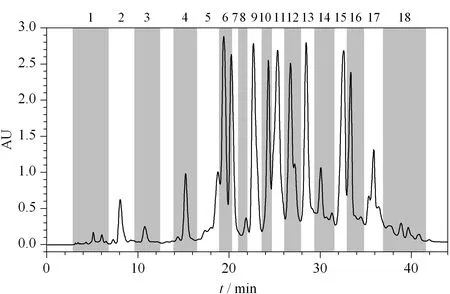
图 3 巴天酸模根乙酸乙酯萃取液一维分离的色谱图Fig. 3 First dimensional chromatogram of ethyl acetate extract of root of Rumex patientia L. Column: phenyl/tetrazolium (250 mm×4.6 mm, 7 μm). Mobile phases: (A) 0.1% (v/v) formic acid aqueous solution and (B) methanol. Gradient elution: 0-5 min, 25%B; 5-50 min, 25%B-90%B; 50-55 min, 90%B. Detection wavelength: 266 nm. Injection volume: 30 μL. Flow rate: 1. 0 mL/min.

图 4 巴天酸模根一维色谱10个流分的二维分离色谱图Fig. 4 Second dimensional chromatograms of ten fractions abstracted from the root of Rumex patientia L. with first dimensional chromatography Column: Unitary C18 (250 mm×4.6 mm, 7 μm). Mobile phases: (A) 0.1% (v/v) formic acid aqueous solution and (B) methanol. Gradient elution: 0-5 min, 25%B; 5-50 min, 25%B-90%B; 50-55 min, 90%B. Detection wavelength: 210-400 nm. Injection volume: 20 μL. Flow rate: 1. 0 mL/min.
2.4 一维流分的二维色谱分析
为了对分析对象进行更加全面的分析,选择二维色谱分离系统对一维制备得到的F1~F18流分进行进一步的分离分析,部分结果见图4。可以看出,一维流分F2、F3、F5、F6、F8、F9、F10、F11、F12和F16在进一步的二维色谱分析中均得到多个色谱峰,有效地增加了巴天酸模根液相色谱分离的峰容量。其中,F6、F9、F10和F12这4个流分均得到较纯的化合物色谱峰(如图4虚线标识所示),可以进行制备得到纯度较高的标准品。
3 结论
本文构建了离线二维反相/反相液相色谱分离系统,实现了对巴天酸模根乙酸乙酯提取液化学成分的有效分离,并有望得到高纯度的标准品。本实验提供的二维分离方法高效、可行,为巴天酸模药材的微量组分的分离和活性化合物的筛选提供了技术支撑。
[1] Pan Z R, Liang H L, Liang C H, et al. Chinese Journal of Chromatography, 2015, 33(1): 22
潘智然, 梁海龙, 梁朝晖, 等. 色谱, 2015, 33(1): 22
[2] Giddings J C. J Chromatogr A, 1995, 703: 3
[3] Zhou D Y, Xu Q, Xue X Y, et al. J Pharmaceut Biomed, 2009, 49(2): 207
[4] Yu X, Xin Y J, Jiang X, et al. Henan Science & Technology, 2016(9): 132
于雪, 辛莹娟, 蒋绪, 等. 河南科技, 2016(9): 132
[5] Xie X M, Sun W Y, Huang J Y, et al. Analytical Chemistry, 2016, 44(7): 1140
谢秀满, 孙万阳, 黄竞怡, 等. 分析化学, 2016, 44(7): 1140
[6] Shen B J, Qin K M, Liu Q D. et al. Scientia Sinica Chim, 2013, 43(11): 1480
沈保家, 秦昆明, 刘启迪, 等. 中国科学: 化学, 2013, 43(11): 1480
[7] Gao W, Song H P, Yang H, et al. Chinese Journal of Chromatography, 2017, 35(1): 121
高雯, 宋慧鹏, 杨华, 等. 色谱, 2017, 35(1): 121
[8] Huang J Y, Tong L, Ding L. Progress in Pharmaceutical Sciences, 2015, 39(5): 357
黄竞怡, 佟玲, 丁黎. 药学进展, 2015, 39(5): 357
[9] Jin H L, Liu Y F, Feng J T. J Sep Sci, 2013, 36(15): 2414
[10] Fan Y P, Fu Y H, Fu Q, et al. J Sep Sci, 2016, 39(14): 2710
[11] Jia Y M, Cai J F, Xin H X, et al. Chinese Journal of Chromatography, 2017, 35(6): 650
贾有梅, 蔡剑锋, 辛华夏, 等. 色谱, 2017, 35(6): 650
[12] Li K, Zhu W, Fu Q, et al. Analyst, 2013, 138(11): 3313
[13] Guo X J, Zhang X L, Feng J T, et al. Anal Bioanal Chem, 2013, 405(10): 3414
[14] An R, Xiao Y. Modernization of Traditional Chinese Medicine and Meteria Medic- World Science and Technology, 2014, 16(3): 549
安蓉, 肖尧. 世界科学技术- 中医药现代化, 2014, 16(3): 549
[15] Xie Z W. National Compendium of Chinese Herbal Medicine. 2nded. Beijing: People’s Medical Publishing House, 1996
谢宗万. 全国中草药汇编. 2版. 北京: 人民卫生出版社, 1996
[16] Laight D W, Carrier M J, Anggard E E. Cardiovasc Res, 2000, 47(3): 457
[17] Liu J, Xia Z T, Zhou G R, et al. Journal of Chinese Medicinal Material, 2011, 34(6): 893
刘景, 夏忠庭, 周桂荣, 等. 中药材, 2011, 34(6): 893
猜你喜欢
广州化工(2022年2期)2023-01-15
大电机技术(2022年5期)2022-11-17
防爆电机(2021年4期)2021-07-28
中国现代中药(2021年4期)2021-06-11
中成药(2021年9期)2021-03-29
中成药(2021年6期)2021-03-29
铁道通信信号(2020年6期)2020-09-21
世界农药(2019年4期)2019-12-30
铁道通信信号(2019年3期)2019-04-25
中成药(2018年2期)2018-05-09
