
低温分配固液萃取结合高效液相色谱-质谱法测定食用菌中6-苄基腺嘌呤
2018-01-11 07:40渡边正树王大利李永路
色谱 2018年1期
侯 琨, 渡边正树, 王大利, 韩 晶, 李永路, 霍 虹
(1. 日立仪器(大连)有限公司, 辽宁 大连 116033; 2. 株式会社日立高新技术科学, 日本 茨城 3120033; 3. 大连市儿童医院, 辽宁 大连 116012; 4. 中国科学院大连化学物理研究所, 辽宁 大连 116023)
食用菌因味道鲜美、营养丰富、药食同源而备受关注。食用菌的蛋白质含量为27% ~48% ,碳水化合物约60% ,脂质在2% ~8%之间,同时含有多种维生素和微量元素[1]。近代医学表明,食用菌除了营养丰富,还有增强机体免疫力、抗肿瘤等功效[2,3]。如营养学家预言,食用菌已成为新的食物资源,全世界有20个品种的食用菌在进行产业化生产[1]。为了提高产量,满足人们对食用菌的大量需求,食用菌生产过程中需要添加植物生长激素[4,5]。
6- 苄基腺嘌呤(6- benzylaminopurine, 6- BA)是人工合成的细胞分裂素,正常剂量下能促进细胞的分裂和生长,具有调运氨基酸、生长素、无机盐等多种效能,对植物的生长、发育有着促进与调节作用[6-8],但过多摄入会刺激人体皮肤黏膜,造成食道胃黏膜损伤,出现恶心呕吐等现象,甚至致癌、致畸[9]。欧洲食品安全局(EFSA)规定[10]植物来源的食品中6- 苄基腺嘌呤最大残留量(MRL)为0.01 mg/kg。GB 2760- 1996规定在豆芽生产中的最大使用量为10 mg/kg,最大残留量为2.0 mg/kg。2015年,中国食品药品监督管理总局农业部中国卫生和计划生育委员会第11号公告已经禁止在豆芽生产中使用6- 苄基腺嘌呤[11]。法律法规多针对“毒豆芽”事件而展开,对于食用菌培育中6- BA的使用导致的残留问题目前国内尚无报道。
6- BA的测定方法主要有酶联免疫法(ELISA)[12]、气相色谱法(GC)[13]、气相色谱- 质谱法(GC- MS)[14]、高效液相色谱法(HPLC)[15]和高效液相色谱- 质谱法(HPLC- MS)[16-19]等。这些方法足以解决6- BA的分离和检测,但样品基质复杂和目标物含量低使得样品前处理成为决定分析结果的关键环节。目前常用的样品前处理方法主要有固相萃取法[20],但成本相对较高,且食用菌样品容易堵塞固相萃取柱,应用此法耗时耗力。因此,开发操作简单、提取分离效率高、高通量、成本低、准确性高的前处理技术用于食用菌中6- BA的测定显得尤为重要。低温分配固液萃取(SLE- LTP)技术能在短时间内同时完成提取和分离,实验步骤大大简化,和固相萃取法相比具有简单、经济的优势[21,22],尤其适合食用菌类复杂生物基质中高极性杂质的去除。经过SLE- LTP前处理,样品可以直接进行检测。
本研究采用SLE- LTP技术,以食用菌中的6- 苄基腺嘌呤为检测目标,采用HPLC进行测定,并采用质谱对结果进行进一步确认,建立了简便、快捷、实用的测定食用菌中6- 苄基腺嘌呤的分析方法。
1 实验部分
1.1 仪器与试剂
Chromaster高效液相色谱仪(配有Chromaster 5110输液泵、5210自动进样器、5310柱温箱、5410紫外检测器)、Chromaster 5610质谱检测器、CF16RXⅡ型高速离心机(日本日立公司); FU- 9H型超声波清洗仪(日本TGK公司); MDF- U338- C型医用低温箱(日本SANYO公司); Milli- Q超纯水器(美国Millipore公司)。
6- 苄基腺嘌呤标准品(纯度大于99.0% ,德国Dr. Ehrenstorfer公司);乙腈、甲醇(色谱纯,美国Tedia公司);乙酸(色谱纯,德国CNW公司);乙酸铵(纯度为95.0% ,日本Wako公司)。各种食用菌样品均购自大连超市。
1.2 标准溶液的配制
准确称取6- 苄基腺嘌呤标准品10 mg,用甲醇溶解并定容至100 mL,配制成质量浓度为0.1 g/L的标准储备液,于4 ℃冰箱中保存。准确移取适量6- 苄基腺嘌呤标准储备液,用甲醇稀释并配制适当浓度的标准工作液。
1.3 样品前处理
新鲜的食用菌样品经高速万能粉碎机以24 000 r/min粉碎20 s。准确称取4.0 g混合均匀的样品,置于15 mL离心管中,加入5 mL乙腈,超声提取15 min,以12 000 r/min的速率离心10 min,取上清液,残渣加入5 mL乙腈,重复上述超声提取和离心操作,合并上清液,向其中加入0.5 mL超纯水,混匀,于-30 ℃医用低温箱中保存,待低温分层4 h后,立即取出上层澄清液,在常温下氮吹至4 mL以下,用乙腈定容至4 mL,过0.45 μm有机滤膜,待分析。
1.4 分析条件
1.4.1色谱条件
色谱柱:Hitachi LaChrom C18色谱柱(250 mm×4.6 mm, 5 μm);柱温:45 ℃;流动相:0.02 mol/L乙酸铵(含0.1%(v/v)冰乙酸)- 甲醇(6∶4, v/v);等度洗脱;流速:1.0 mL/min;进样量:10 μL;检测波长:267 nm。
1.4.2质谱条件
电离方式:电喷雾离子(ESI)源,正离子模式;离子源温度:70 ℃;检测模式:选择离子监测(SIM)模式;离子化电压:2 500 V;气体(N2)流速:0.6 L/min;锥孔电压1:80 V;锥孔电压2:30 V。
2 结果与讨论
2.1 低温分配固液萃取条件的优化
2.1.1提取溶剂的选择
低温分配固液萃取技术是基于低温下有机相和水相分层而引起分析物的分配。分配后食用菌样品中的多糖类物质及其他高极性杂质被冻结在水相中,而目标物溶解在有机溶剂中,进而达到净化的目的。乙腈、甲醇、乙酸乙酯是提取6- 苄基腺嘌呤时最常用的3种溶剂,本实验采用这3种溶剂进行提取- 低温分配效果的考察。结果表明:在常温、低温(-30 ℃)及超低温(-80 ℃)下,以甲醇作提取剂的溶液体系直至冻结也没有获得两相分离,即没有低温分配,因此不适合作为本实验的提取溶剂;乙酸乙酯作为提取剂时目标物的回收率不高,在24% ~32%之间;以乙腈作提取剂的溶液体系在-30 ℃下保持12 h,通过液液分配,分析物可转移到有机相,高极性的杂质停留在水相,利用低温分配后,提取的绝大部分杂质都能被去除,目标物的回收率为48% ~81% ,从而确定乙腈为低温分配的提取溶剂。
2.1.2提取体积的选择
考察了乙腈体积(6、 8、 10、 12和14 mL,分别平均分为2份,提取2次后合并提取液)对目标物回收率的影响。从图1可以看出,随着乙腈体积的增加,6- 苄基腺嘌呤的回收率逐渐增大,当乙腈体积大于10 mL时6- BA的回收率变化平缓,因此,实验采用10 mL乙腈(分2次提取,每次提取体积为5 mL)进行提取。

图 1 提取溶剂体积对6- 苄基腺嘌呤回收率的影响(n=6)Fig. 1 Effects of volume of extraction solvent on the recoveries of the 6- benzylaminopurine (6- BA) (n=6)
2.1.3辅助液体积的选择
低温分配前在溶液体系中加入辅助液(水)可以促进极性杂质向水相移动以获得更纯净的分析样品。本研究考察了水的体积(0、0.5、1.0、2.0和4.0 mL)对目标物回收率的影响。由图2可知,水的体积和6- BA的回收率呈负相关,综合考虑回收率及去除杂质的效果,将水的体积确定为0.5 mL。
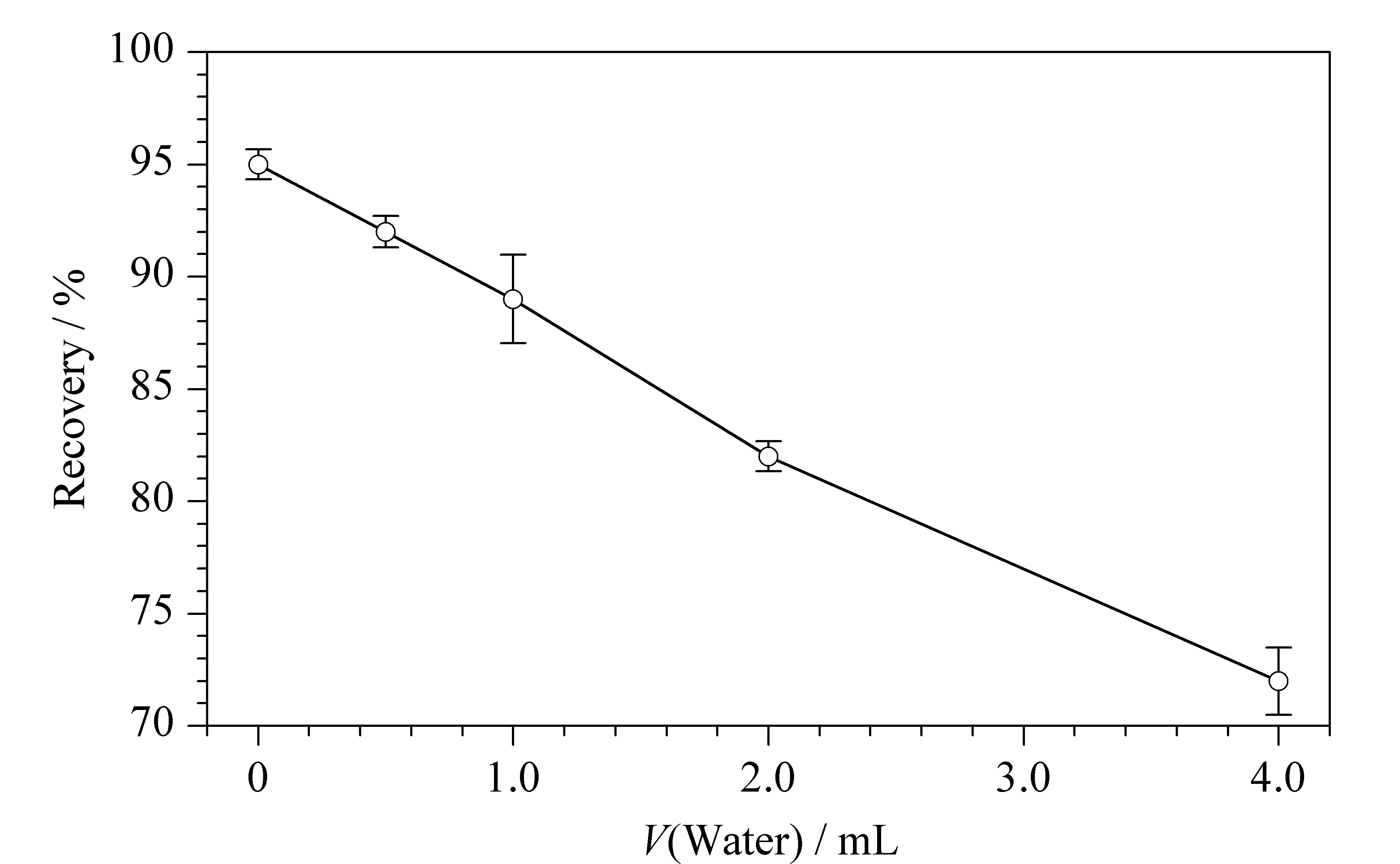
图 2 辅助液体积对6- 苄基腺嘌呤回收率的影响(n=6)Fig. 2 Effects of volume of auxiliary solution on the recoveries of the 6- BA (n=6)
2.1.4低温分配时间的选择
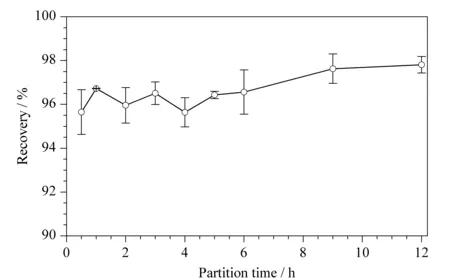
图 3 低温分配时间对6- 苄基腺嘌呤回收率的影响(n=6)Fig. 3 Effects of low- temperature partition time on the recoveries of the 6- BA (n=6)
低温分配需要一个低温静置的时间,为了减少静置时间,对两相的分离动力学进行评价。对低温静置时间进行考察,在0~12 h之间,6- BA的回收率均稳定在95%左右(见图3)。低温时,液相开始分离,0.5~2.0 h后液相保持着稳定的两相分层状态;3 h后水相可见少量冰碴;4 h左右,约80%的水相被冷冻,其分层界面较3 h前更清晰;5 h后水相被全部冷冻,乙腈层开始出现冰碴,水相与乙腈相的分界线变模糊;6 h后水相呈白色固相,水相与乙腈分界线变更模糊。从实验数据可以发现,水相冻实后将阻断水相中物质的分配和转移。因此,推测物质的分配与转移发生在水相冰冻之前。综合考虑,选择低温分配的静置时间为4 h。
2.2 色谱条件的优化
[19]的方法,比较了0.02 mol/L乙酸铵(含0.1%(v/v)冰乙酸)与甲醇在不同体积比所形成的流行相条件下对目标化合物分离度和峰形的影响。随着甲醇体积分数的增加(由35%增至50% ), 6- 苄基腺嘌呤在固定相上的保留减弱,为了避免杂质峰的干扰,选择0.02 mol/L乙酸铵(含0.1%(v/v)冰乙酸)- 甲醇(6∶4, v/v)作流动相,6- 苄基腺嘌呤的保留时间为16.4 min。
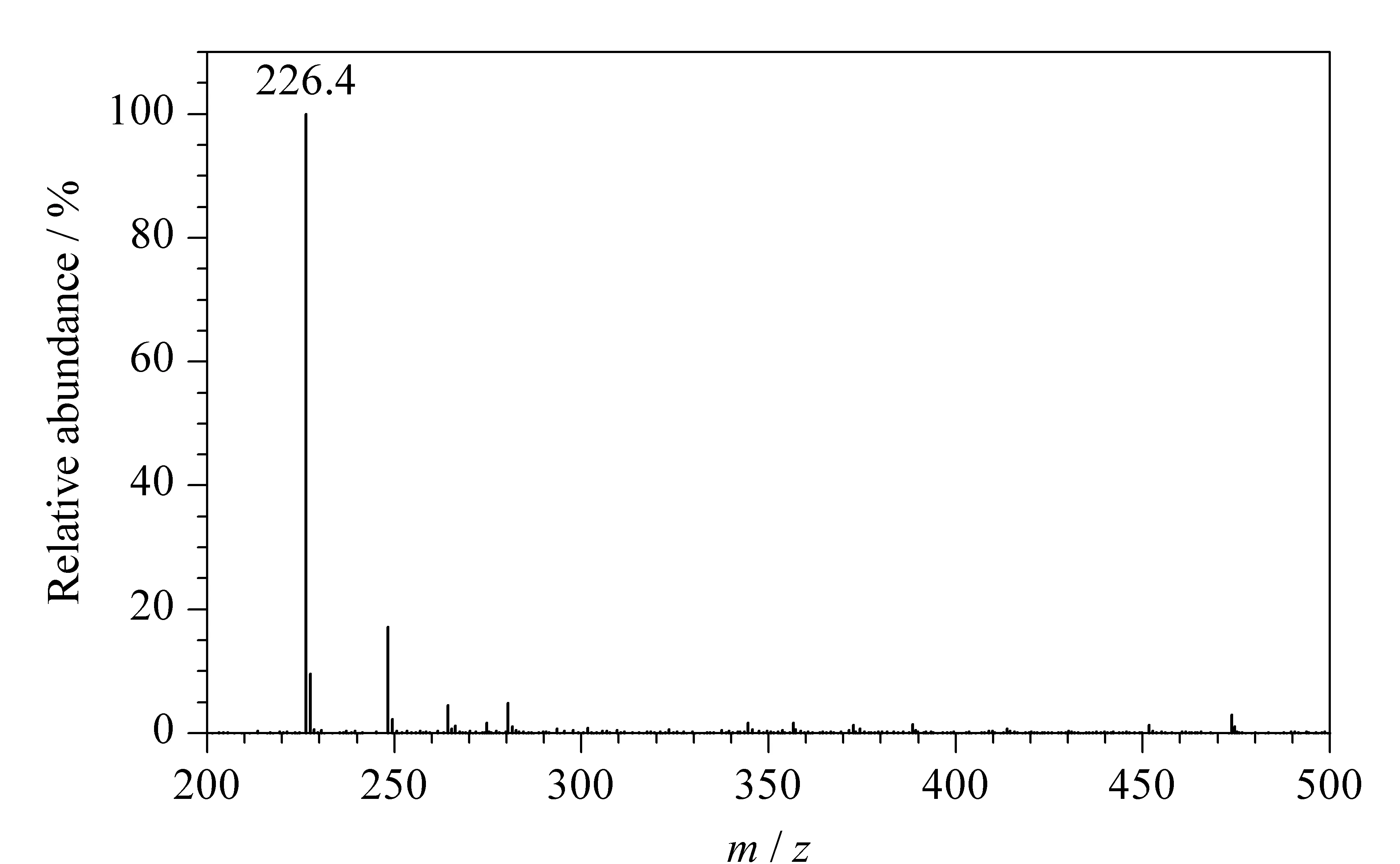
图 4 6- 苄基腺嘌呤的质谱图Fig. 4 Mass spectrum of 6- BA
2.3 质谱条件的优化
6- 苄基腺嘌呤的结构中含有氨基,在正离子模式下更易得到较强的准分子离子信号。本实验选择ESI+模式,采用注射泵直接连续进样,以2.0 μL/min的速率注入10 mg/L的6- 苄基腺嘌呤标准溶液,全扫描条件下获得6- 苄基腺嘌呤的准分子离子峰(见图4),并对此离子(m/z226.4)进行SIM模式扫描。
2.4 线性范围、检出限和定量限
将1.2节中配制的不同质量浓度(0.05、0.1、0.2、0.5、1.0和2.0 mg/L)的标准工作液重复分析3次,以目标物峰面积(Y)对其在标准工作液中的质量浓度(X, mg/L)进行线性回归。以3倍和10倍信噪比(S/N)对应的目标物质量浓度为检出限(LOD)和定量限(LOQ)。6- 苄基腺嘌呤的线性方程、相关系数(r2),线性范围、检出限和定量限见表1。

表 1 6-苄基腺嘌呤的线性方程、相关系数、线性范围、检出限和定量限
Y: peak area;X: mass concentration, mg/L.
2.5 回收率和精密度
在4 g(精确到0.01 g)香菇等7种食用菌样品中进行加标回收试验,每个基质的加标水平分别为0.1 mg/kg和0.5 mg/kg,每个水平平行测定6次,计算加标回收率和相对标准偏差(RSD),结果见表2。7种基质中6- 苄基腺嘌呤的平均加标回收率为81.3% ~93.7% ,相对标准偏差为0.7% ~2.4% 。
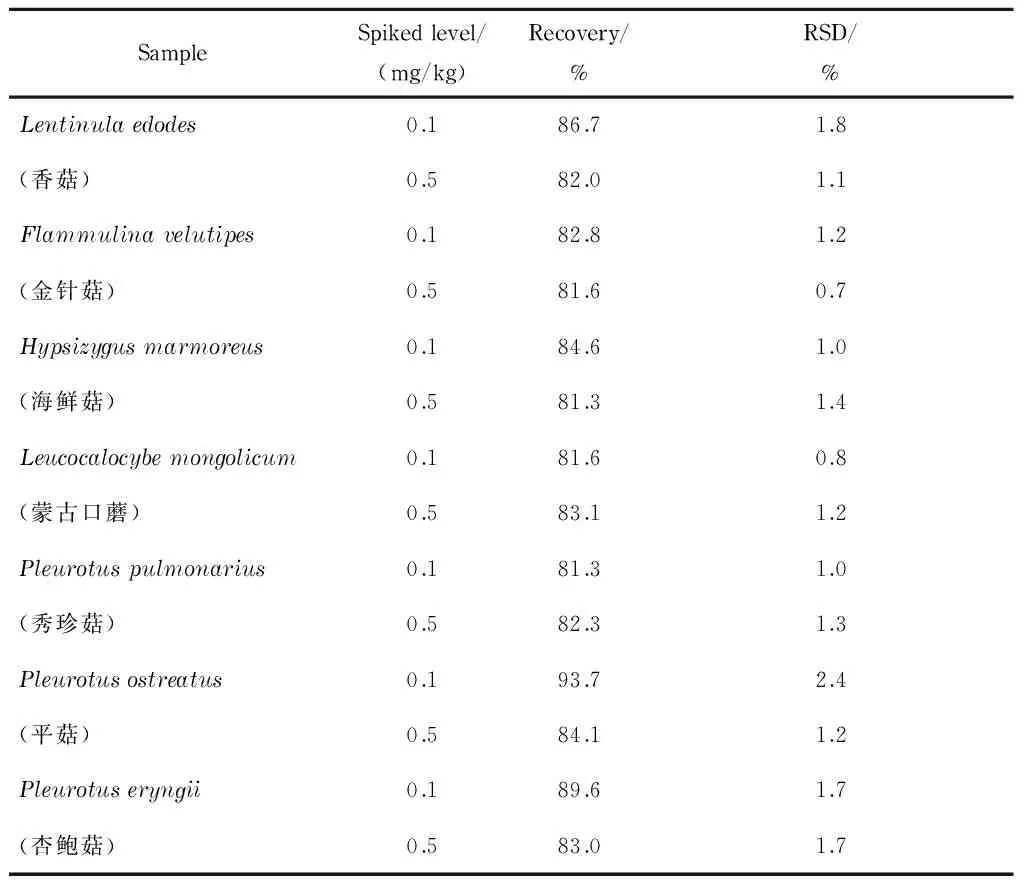
表 2 7种蘑菇样品中6-苄基腺嘌呤的加标回收率和相对标准偏差(n=6)
2.6 实际样品分析
使用本方法对从大连市场购买的24份食用菌样品进行测定,全部样品中均未检出6- 苄基腺嘌呤。用质谱仪在SIM模式下对食用菌样品及加标样品进行确认,食用菌样品中不含有6- 苄基腺嘌呤。杏鲍菇实际样品和定量限添加水平下的杏鲍菇实际样品的色谱图见图5。

图 5 (a)杏鲍菇实际样品和(b)杏鲍菇加标(0.5 mg/kg)样品的色谱图Fig. 5 Chromatograms of (a) a pleurotus eryngii sample and (b) a pleurotus eryngii sample spiked with 0.5 mg/kg 6- BA
3 结论
本文建立了食用菌样品中6- 苄基腺嘌呤的检测方法,样品经乙腈提取,低温分配固液萃取,高效液相色谱- 质谱法测定。该法前处理步骤简单、快速,大大降低了前处理的成本。方法评价结果表明,该法准确、可靠、能够满足国内关于6- 苄基腺嘌呤残留限量的要求,同时也为其他种类食品中6- 苄基腺嘌呤的检测提供了新思路。
参考文献:
[1] Sanchez C. Appl Microbiol Biot, 2004, 64(6): 756
[2] Greeshma P, Ravikumar K S, Neethu M N, et al. Int J Med Mushrooms, 2016, 18(3): 235
[3] Kashina S, Flores Villavicencio L L, Zaina S, et al. Int J Med Mushrooms, 2016, 18(1): 49
[4] Qiu C E, Tu J M, Wang W D, et al. Journal of Microbiology, 2001, 21(3): 63
邱昌恩, 涂俊铭, 王卫东, 等. 微生物学杂志, 2001, 21(3): 63
[5] Wang Q, Liu M, Xu X C, et al. Journal of Hebei University (Natural Science Edition), 2012, 32(3): 286
王谦, 刘敏, 徐啸晨, 等. 河北大学学报(自然科学版), 2012, 32(3): 286
[6] Sun D, Zhang H J. Anal Chim Acta, 2006, 557(1/2): 64
[7] Ohashi F, Ueda S, Taguri T, et al. Appl Clay Sci, 2009, 46(3): 296
[8] Pan B Z, Xu Z F. J Plant Growth Regul, 2011, 30(2): 166
[9] United States Environmental Protection Agency (US EPA). The US EPA Pesticide Toxicity Database. (2009- 10- 19) [2016- 08- 22]. http://www3.epa.gov/pesticides/chem_search/ppls/072639- 00003- 20091019.pdf
[10] European Food Safety Authority (EFSA). EFSA J, 2010, 8(10): 1716
[11] China Food and Drug Administration. 2015 Years No. 11 Bulletin of the China Food and Drug Administration. (2015- 04- 13) [2017- 11- 24]. http://www.sda.gov.cn/WS01/CL0050/118260.html
国家食品药品监督管理总局. 国家食品药品监督管理总局公告2015年第11号. (2015- 04- 13) [2017- 11- 24]. http://www.sda.gov.cn/WS01/CL0050/118260.html
[12] Zhang W, He L S, Zhang R, et al. Food Chem, 2016, 207: 233
[13] Lee S M, Kim J Y, Lee H J, et al. J Korean Soc Appl Bi, 2014, 57(1): 83
[14] Wu P G, Tan Y, Zhang J, et al. Chinese Journal of Analytical Chemistry, 2014, 42(6): 866
吴平谷, 谭莹, 张晶, 等. 分析化学, 2014, 42(6): 866
[15] Ma X F, Chen G, Qin D P, et al. Agrochemicals, 2016, 55(2): 124
马雪丰, 陈果, 秦德萍, 等. 农药, 2016, 55(2): 124
[16] Kim K G, Park D W, Kang G R, et al. Food Chem, 2016, 208: 239
[17] Xie H B, Zhou M Y, Zhao H F, et al. Chinese Journal of Chromatography, 2014, 32(5): 493
谢寒冰, 周明莹, 赵海峰, 等. 色谱, 2014, 32(5): 493
[18] Liu H, Wu B, Yin Y, et al. Chinese Journal of Chromatography, 2013, 31(1): 22
柳菡, 吴斌, 殷耀, 等. 色谱, 2013, 31(1): 22
[19] Zhang J, Wan H H, Zhang H. Chinese Journal of Chromatography, 2017, 35(9): 963
张婧, 万慧慧, 张华. 色谱, 2017, 35(9): 963
[20] GB/T 23381- 2009
[21] Sousa D A, Goncalves R M, Heleno F F, et al. Microchem J, 2014, 114: 266
[22] Costa A I G, Queiroz M E L R, Neves A A, et al. Food Chem, 2015, 181: 64
猜你喜欢
农业工程学报(2022年5期)2022-06-22
世界科学技术-中医药现代化(2021年5期)2021-11-05
理化检验-化学分册(2020年5期)2020-06-15
石油地质与工程(2019年3期)2019-09-10
中国钼业(2019年2期)2019-01-19
采矿技术(2016年2期)2016-12-14
合成化学(2015年10期)2016-01-17
合成化学(2015年1期)2016-01-17
中国洗涤用品工业(2015年9期)2015-02-28
分析化学(2014年8期)2014-09-02
