
基于病例对照组的检验差异甲基化基因功能区域的方法
2018-01-10 11:11:59马欣宇胡跃清
复旦学报(自然科学版) 2017年6期
马欣宇,王 婵,胡跃清
(复旦大学 生命科学学院 生物统计学与计算生物学系,上海 200433)
基于病例对照组的检验差异甲基化基因功能区域的方法
马欣宇,王 婵,胡跃清
(复旦大学 生命科学学院 生物统计学与计算生物学系,上海 200433)
DNA甲基化是一种重要的表观遗传学修饰,能够参与基因表达调控从而影响生理活动.在病例-对照组研究中,利用甲基化水平定位与疾病相关联的基因功能区域,有助于了解甲基化调控基因表达的机制,为后续研究提供疾病关联基因的线索.目前检验甲基化差异区域的方法存在一定不足: 一是缺乏定义差异甲基化区域的统一标准;二是检测结果存在横跨多个基因功能区域的现象,难以进行生物学解释.本文基于得分检验(Z-score test)提出了以基因功能区域为单位的检验差异甲基化水平的方法cZST.该方法的特点是依靠位点物理距离和染色体全局信息修正协方差矩阵.模拟研究表明cZST在亚硫酸氢盐测序数据样本量小、读数低、扰动位点多的情形下具有高功效,且优于现有方法.将cZST运用到乳腺导管癌真实数据中,检测出与疾病显著相关的661个基因功能区和81个单位点.基因注释分析说明了cZST方法的有效性,并发现Dbl、Pleckstrin基因家族及Rho调控基因的甲基化与乳腺导管癌紧密相关,为后续研究提供指引.
差异甲基化检验; 亚硫酸氢盐测序; 功能富集分析
DNA甲基化是发生在生物DNA链核苷酸上的一种化学修饰.在真核细胞中,S-腺苷甲硫氨酸上的甲基会在DNA甲基转移酶的作用下,转移到DNA分子CpG序列的胞嘧啶上[1].人类DNA中约70%~90%CpG二核苷酸存在甲基化修饰[2],并且甲基化的CpG二核苷酸大部分聚集在基因的5’端,称为CpG岛,长度在几百至一千个碱基对(bp)左右,它们大多分布在基因的上游、启动子和外显子区域中[3].
研究表明CpG岛上的甲基化会显著影响基因的转录调节,通常会使转录失活.例如,在人和啮齿类动物中,病毒序列可以与插入的基因交联而甲基化,从而使该基因沉默[4].DNA甲基化修饰是可逆的,并且不直接依赖于序列,能够参与基因表达调控[5-6].因此,DNA甲基化作为研究人类疾病、发育、衰老的重要媒介,已成为当今基因组学的重要研究对象之一.
第二代测序技术,由于其高灵敏度和特异性,已经成为表观基因组研究的有力工具.BS-seq(Bisulfite Sequencing)使用亚硫酸氢盐处理DNA,未甲基化的胞嘧啶会转化为尿嘧啶,而甲基化的胞嘧啶不发生变化[7].因此,BS-seq能获得基因组的全碱基甲基化信息,从而计算出单核苷酸分辨率下甲基化在细胞样本中的发生率,即甲基化水平值,它等于被甲基化信号强度与总信号强度之比.由于全基因组BS-seq较为昂贵,许多研究者使用简易BS-seq(RRBS),它基因组覆盖率不到10%,测序成本相对较低[8-9].
随着芯片技术和高通量测序技术的发展,全表观基因组关联研究成为一个研究热点.它通过检验基因组上,尤其是CpG岛上,DNA核苷酸甲基化的分布差异,搜索与性状关联性强的位点或区域.已有的方法包括COHCAP[10]、BISEQ[11]、HMM-Fisher[12]等,它们能够较为准确地检测BS-seq单位点的甲基化差异,但存在一定的局限性.由于多数CpG位点成簇存在,共同影响所在基因,而一个基因功能区域内各甲基化位点的表达调控作用是互补累加的,因此单位点差异甲基化检验提供的信息往往是不充分甚至片面的.而针对连续多个CpG位点的差异甲基化区域检验,缺乏划分或拼接区域的统一标准[13],难以比较和重复验证.同时其分析结果常常横跨多个功能区域甚至两个基因,这给进行生物学解释带来了困难.
本文提出一种基于得分的差异甲基化检验,它考虑了差异的方向性,量化了基因功能区域的差异显著程度,其生物学意义明显,而且方便后续研究.同时针对甲基化BS-seq数据样本量较小、测序读数低的特点,提出了依靠位点物理距离和染色体全局信息修正协方差矩阵的策略,它显著地提高了检验功效(真阳性率,TPR).
1 符号与方法


(1)
在原假设H0下,即病例组和对照组的甲基化水平一致时,U近似服从均值为0,协方差矩阵为V的多维正态分布,从而构造检验统计量S=UTV-1U,其中
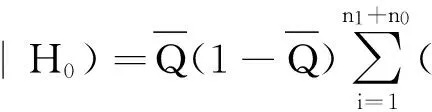
(2)
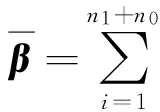
由于测序成本高,BS-seq数据样本量小,测序读数低.而低读数位点会导致甲基化水平的估计值误差较大,从而协方差偏差也较大,进而影响检验功效.为了弥补这种不足,我们引入全局甲基化信息,利用同一染色体上其他位点甲基化水平的相关系数,来校正低读数位点协方差的估计,从而增加检验功效.
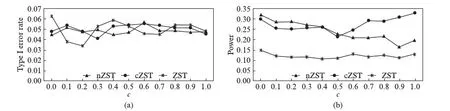
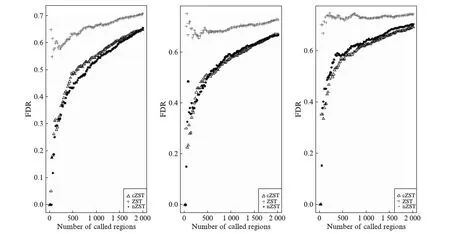
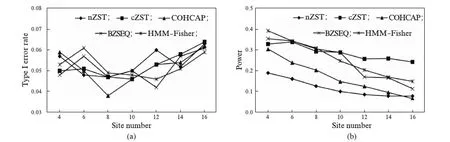
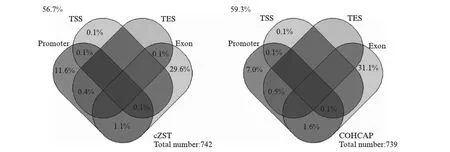
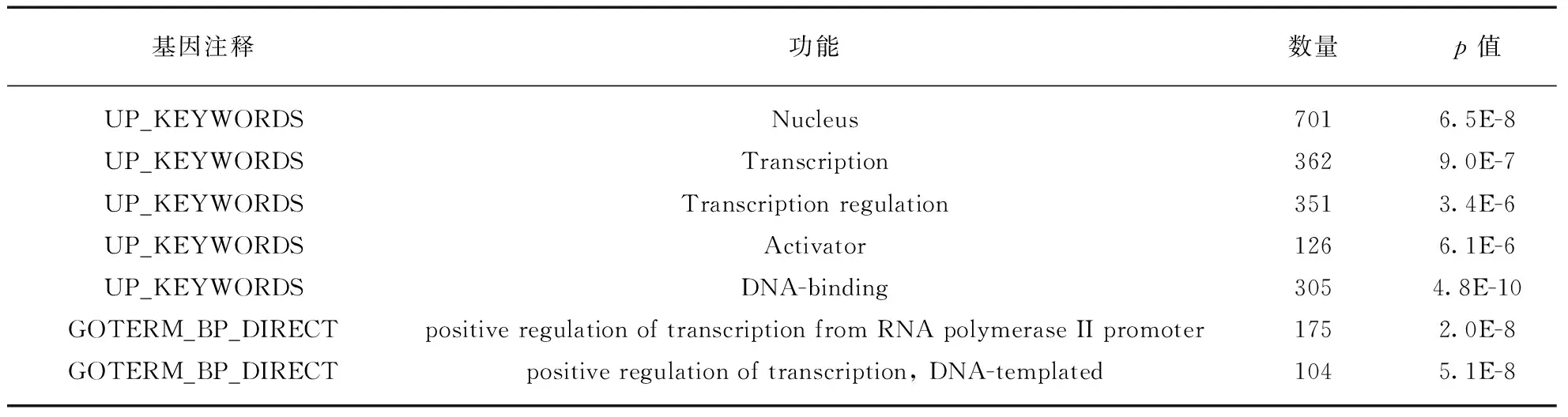
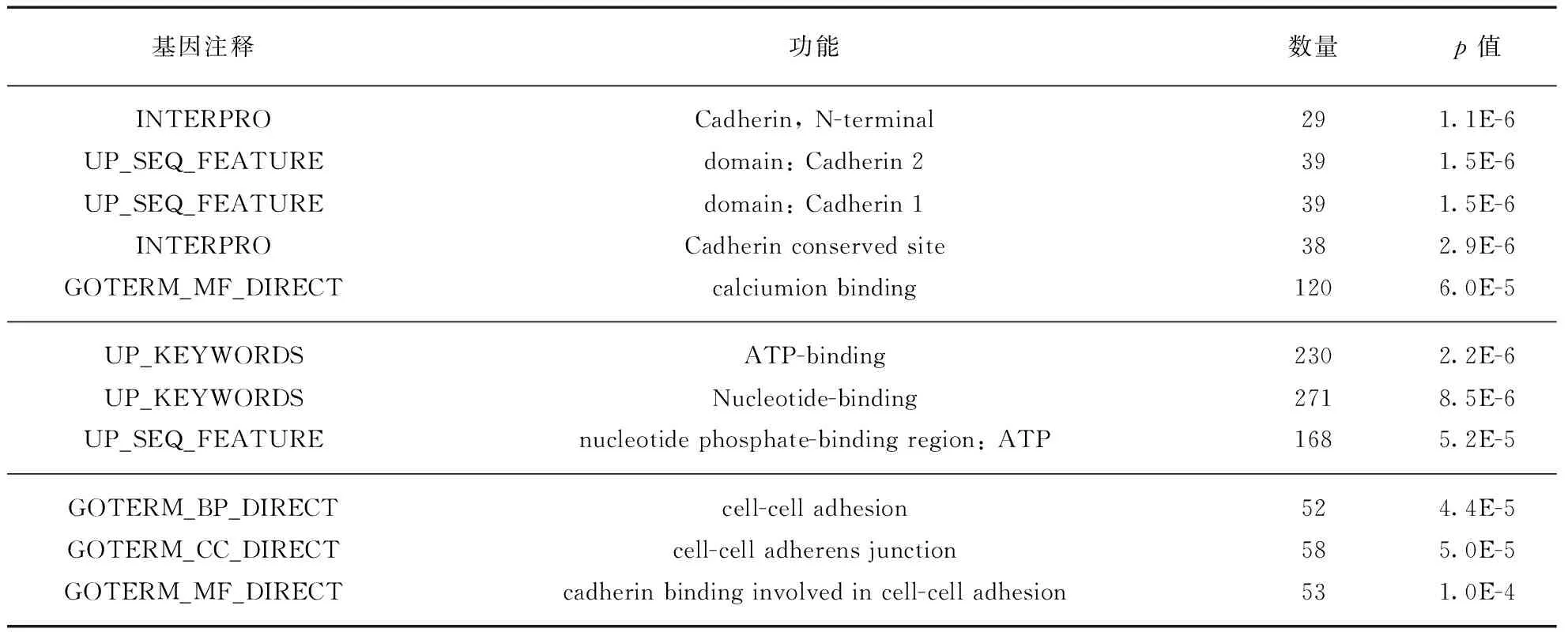
具体校正策略如下.先计算整条染色体上两两位点的物理间距以及相应的甲基化水平的相关系数(Correlation coefficient),并进行局部加权线性回归获得LOWESS曲线,作为全局信息以备后面使用.然后设定两个读数阈值T1=12和T2=4,对位点进行分类.给定某位点,记T为所有个体的总信号读数(甲基化和非甲基化信号之和)的平均值.若T≤T2,则认为此位点的数据质量低,将其从数据集中剔除;如果T2 该修正策略基于甲基化BS-seq数据如下的特征: 同一条染色体上相同物理距离的两位点甲基化水平的相关系数的波动程度小,因此这些相关系数的平均值与低读数位点真实相关系数较为接近.我们选用一个RRBS真实数据集[15],对1号染色体上位点间甲基化水平相关系数进行统计分析,其平均值及变异系数(Coefficient of Variation, CV)相对物理距离的散点图见图1.我们发现在点间距小于一定范围(如400bp)的两位点对应β值相关系数明显大于0且与间距呈反比,并且当间距近时,同一物理距离的两位点β值相关系数的变异系数很低,说明相关系数波动程度小,这说明了修正策略的合理性. 此外,如果忽略任意两位点之间的甲基化水平的相关性,即相当于令V的非对角元素全部为零,对角元为样本方差,这种得分检验记作nZST(non-related ZST),将它与cZST、ZST一起进行比较. 统计量S的p值可采取置换[16]来获取.在每次置换时,我们随机打乱每个个体的患病状态,然后计算新统计量Sm,1≤m≤M.则p值为 (3) 在模拟研究中,基于R次重复模拟数据的p值pr,r=1,2,…,R,在显著性水平α下,该方法的功效RTP为 (4) 图1 β值相关系数的均值和变异系数散点图Fig.1 Mean value and CV of β values’ correlation coefficient(a) RRBS数据1号染色体上同距离两位点的β值相关系数的均值随物理间距的变化情况;(b) 相关系数的变异系数随物理间距的变化情况.实线为LOWESS拟合平滑曲线. 为了模拟实际BS-seq数据特征,我们通过多维正态分布刻画位点间甲基化水平相关性,通过二项分布模拟产生测序信号.假设某区域的总长度为800bp,含L个CpG位点,随机从1到800中选取L个不同的位置记为D1,D2,…,DL.考虑两个多维正态分布N(μ1,Σ)和N(μ0,Σ),μ1、μ0为两个均值向量,对应协方差矩阵Σ=(σil)L×L,其中的对角元均为1,非对角元为 σil=exp(-dil/d0), (5) 其中dil=|Di-Dl|,d0为默认任意两位点β值存在相关性的距离参数,在我们的随机模拟中取d0=400bp.该随机模型建立了物理距离与位点相关性之间的联系,距离更近的两个位点甲基化水平拥有更大的相关系数,反之亦然. 以多维正态分布N(μ1,Σ)或N(μ0,Σ)产生随机数,通过逆Logistic变换得到取值在[0,1]的L个值P1,P2,…,PL,作为观察到甲基化信号的理论概率;设单位点测序总读数的期望均为T0,通过泊松分布P(T0)产生L个随机数N1,N2,…,NL,作为测序总读数.最终通过二项分布B(Nl,Pl)产生L个位点上的甲基化和未甲基化读数,进而得到甲基化水平值β1,β2,…,βL.因此,每次模拟产生L个不同的位置,从N(μ1,Σ)出发可以产生n1个病例的L个甲基化水平值,从N(μ0,Σ)出发可产生n0个对照的L个甲基化水平值.取n1=n0=6. 实验1: 在测序读数足够大且位点存在相关性的条件下,比较ZST、cZST和nZST 3种检验方法的表现.模拟研究中cZST方法全部使用修正的协方差.基于原假设,取μ1=μ0=0,产生4000组无差异数据;基于备择假设,取μ1≠μ0,产生1000组有差异数据.假设这5000组数据对应的区域位于同一染色体上,且两两区域间距较远.分别使用上述3种方法进行检验,将P值从小到大排序,依次选取1到前2000个作为显著性区域,计算错误发现率(False Discovery Rate, FDR). 当T0=25,即测序读数较大时;或令β值完全等于甲基化理论概率,即βi=Pi,i=1,2,…,L时,分别产生模拟数据,FDR结果如图2所示.图2结果表明,利用全局甲基化水平的相关系数信息的cZST控制FDR的能力远强于使用原始协方差矩阵的ZST及不考虑位点相关性的nZST. 图2 3种方法在测序总读数大时FDR比较Fig.2 FDR values of three methods given a large reading depth3种方法选取不同显著位点个数时对应的FDR值.μ0始终为0,原假设下μ1=0,备择假设下μ1=(0,0,0,1,1,1,0,0). 实验2: 当位点之间的β值相关性较弱时,全局甲基化相关系数提供的真实信息减少,cZST可能受到干扰,探究该情况下cZST的稳健性.在模型中加入一个用于调整相关性的权重系数c,将多维正态分布协方差矩阵设为 Σ′=cΣ+(1-c)I,c∈[0,1]. 则c取值越小,位点间模拟β值的相关性越弱.其余参数不变,原假设下产生数据计算3种方法的第一类错误率(type I error rate),备择假设下产生数据比较它们功效的差异,α=0.05,结果如图3所示.在三者均能控制住第一类错误率的前提下,c>0.5时,cZST功效优势持续增加;cZST与nZST在c=0.4~0.5时功效类似,而且二者始终优于ZST.这说明即使β值相关性较弱时,我们提出的协方差修正方法仍有优秀的表现. 图3 3种方法第一类错误率与功效随参数c的变化Fig.3 Type I error and power comparison among three methods under different values of ccZST、ZST、nZST第一类错误率(a)与功效(b)与随参数c值变化情况(α=0.05).3种方法均能将第一类错误率控制在0.05左右.c≥0.4时cZST即可作为首选方法.T0=15,其余参数同实验1. 实验3: 探究ZST、cZST和nZST在测序读数小的数据集上的功效表现,c取0.3令β值相关性较弱,T0分别取15、10和5,3种方法FDR结果见图4.由实验2我们已知,T0=15,c=0.3时,nZST功效优于cZST,能将FDR控制在更低的范围,如图4左图所示.但当T0减小时(图4中、右),同水平下cZST的FDR变得比nZST更低,意味着当区域测序读数降低时,cZST方法将具有格外的优势. 图4 3种方法在测序总读数不同时FDR比较Fig.4 FDR values of three methods under different reading depths模型的泊松分布期望T0变化时cZST、ZST、nZST方法的FDR值.T0=15(左),10(中),5(右).c=0.3,其余参数同实验1. 实验4: 将cZST、nZST与已有方法比较.我们提出的方法能够以基因功能区域为单位,量化甲基化水平差异程度,获得单一p值,供疾病基因关联研究.目前,适用BS-seq数据,在染色体区域上搜索差异甲基化的方法有COHCAP[10]、BISEQ[11]、HMM-Fisher[12]等.它们使用的统计方法、拼接区域的标准不尽相同,方法之间难以互相比较.因此我们采用Dolzhenko等[17]在BS-seq研究中使用的Stouffer-Liptak检验[18],计算单位点检验之间的协方差,通过合并已有方法在基因功能区域内平行的多个DML检验p值,最终得到单一p值,与我们的方法进行功效比较. 我们将功效表现较好的cZST、nZST与上面3种已有方法进行功效比较.区域位点总个数L从4变化到16,令μ1=μ0=0产生数据,计算第一类错误率,再调整μ1中央的两个值为1,即令区域中央2个位点甲基化水平存在差异,比较各方法功效,结果如图5所示.在能够控制住第一类错误率的前提下,cZST检验功效随L增大而缓落,在位点总数超过8个以后表现优于其他所有方法.说明cZST方法是一种灵敏度较高的方法,功效主要取决区域内存在差异甲基化位点的个数和差异情况,受无关干扰位点个数影响较小. 图5 5种方法第一类错误率与统计功效随区域内位点个数变化的趋势Fig.5 Type I error rate and power comparison among methods under different site numbers in a region存在甲基化差异的位点个数设为0(a)或2(b),令L从4变化到16产生模拟数据,分别比较5种方法第一类错误率与功效的变化情况(α=0.05).c=1,T0=15,其他参数同实验1. 综上,针对基因功能区域的cZST检验有着较高的灵敏度,当区域内扰动位点多时,功效优于已有方法.且在两位点β值的相关系数与距离有关的情况下,无论测序平均读数多少,该种策略相比原始ZST,均能在控制住第一类错误率的同时带来功效提升.分析测序平均总读数低的数据时,检验优势更为明显. 我们选用来自一项乳腺导管癌研究的GEO编号为GSE69993[19]的甲基化RRBS数据,应用cZST进行差异甲基化检验.考虑到环境因素对DNA甲基化测序的影响,我们使用同一批次的15个病例数据和5个对照数据.首先,对415985个候选位点进行预处理,丢弃平均读数少于4个的位点.其次,对剩余位点使用TxDb.Hsapiens.UCSC.hg19.knownGene程序注释基因信息,包括基因名称以及是否处于启动子、转录起始位点、外显子及转录终止位点上.利用注释信息我们将位点划归区域,注释信息与前一个位点不同或间距超过1000bp的CpG位点将被划入下一个新区域.此外,为保证检验的可靠性及运算速度,将位点数超过40的区域平均分为两个小区域.对所有区域进行标号,包含多位点区域的数目与包含单位点区域的数目比值约为0.93.最终我们筛选出32360个区域,修正协方差矩阵,并进行多位点得分检验;在对筛选出的34682个单位点使用得分检验时,由于只有一个位点,cZST与nZST是等价的.最后进行104次置换检验,来计算p值.同时,运用COHCAP对这些区域和单位点进行差异甲基化检验,并与cZST结果进行比较. 图6展示了cZST方法的运行结果,图上标示出了5个乳腺癌关联基因的p值水平,这5个基因均出现在Cancer Genetics Web乳腺癌基因集中,并有大量研究表明其与乳腺癌的关联性[20-24],这说明cZST能有效筛选出疾病关联的基因功能区域.当p值的阈值为0.0001时,则cZST检测出672个区域和129个单位点,涉及3560个CpG位点.随后把间距小于1000bp且注释相同的区域和单位点进行合并,得到661个基因功能区和81个单位点,合计742个显著结果.基于同样阈值下,COHCAP检测出2920个显著性CpG位点,利用基因注释信息分为571个区域和168个单位点共739个显著结果.cZST显著性功能区域与单位点个数比值为8.16,远高于待检时的0.93,也高于COHCAP的3.40,说明cZST方法在基因功能区域方面更具优势. 图6 cZST检验结果的曼哈顿图Fig.6 Manhattan plot of cZST test results检验结果显著性水平在各染色体中的分布情况.各位点图中标注了5个乳腺癌关联基因,其基因功能区检验p值均≤10-4. 从检验显著性结果相对CpG岛的位置分类来看,cZST方法76.3%的显著性位点位于CpG岛上,COHCAP则为77.5%,两种方法检验结果都说明大部分甲基化对表达调控是通过CpG岛的甲基化或去甲基化实现的,这一点符合以往研究结论[25].显著性结果基因注释信息如图7所示,cZST方法约43.3%的显著性结果能被hg19基因注释覆盖(维恩图彩色部分),此比例在COHCAP方法中约为40.7%.在外显子和转录起始位点占比上,cZST结果基本与COHCAP的结果持平,启动子占比cZST远高于COHCAP.结合以往研究结论,启动子或转录起始位点发生甲基化对基因表达影响更大,因此甲基化水平差异也应大概率地出现在这两个区域,这从侧面说明cZST结果比COHCAP更为可靠. 图7 cZST与COHCAP显著性结果注释信息维恩图Fig.7 Annotation Venn diagrams of cZST and COHCAP results图中空白部分数字代表没有基因注释标记的显著性结果占比. 将cZST与COHCAP检验p值从小到大排序,取前2000个基因功能区域,使用DAVID工具进行富集分析并聚类功能注释[26],按相似程度整合得到若干功能大类.选取两种方法前4个的功能注释聚类,按显著性水平将注释词条排序,结果见表1、表2. 表1 cZST显著结果基因富集分析功能注释集Tab.1 Functional annotation clusters of gene enrichment analysis with cZST results 注: 表中E-x表示×10-x. 表2 COHCAP显著结果基因富集分析功能注释集Tab.2 Functional annotation clusters of gene enrichment analysis with COHCAP results (续表) 注: 表中E-x表示×10-x. cZST对应富集分析结果前4类基因注释集依次为: PH结构域、基因转录调控、Rho蛋白DH结构域以及钙黏着蛋白;COHCAP对应富集分析结果前4类基因注释集依次为: 基因转录调控、钙黏着蛋白、ATP结合以及细胞黏连.方法在功能注释集: 基因转录调控和钙黏着蛋白词条上有着类似结果,可以相互验证.基因转录调控毋庸置疑是DNA甲基化最重要的生物功能.文献资料表明钙黏着蛋白的表达直接影响肿瘤的浸润和转移[27],已有研究证实了E-黏着蛋白启动子甲基化调节该蛋白表达水平,并与乳腺导管癌患病状态及生存时间关联[28-29].以上两个功能聚类的结果在cZST富集分析中排名第二、第四,这佐证了该方法的效用. 而cZST对应的第一、第三类富集结果指向了小G蛋白Rho调控及影响小G蛋白构型的PH结构域(Pleckstrin homolgy domain)、DH结构域(Dbl homolgy domain)相关基因.已有很多研究探讨了在乳腺导管癌样本中Rho表达与肿瘤转移状况的相关性和可能的生物学机理[30-31].与COHCAP对应结果中第三注释集ATP结合功能、第四类注释集细胞黏连相比,cZST的基因富集功能聚类与乳腺癌关联更大.并且cZST显著性基因富集分析结果说明,Rho调控基因、Pleckstrin及Dbl基因家族的基因功能区域甲基化,可能影响小G蛋白Rho的构型和活性,进而在乳腺导管癌的致病或扩散恶化过程中起作用,对后续研究起到较好的指引作用. 本研究中,我们从观察真实数据出发,基于染色体上同间距甲基化位点的相关系数离散度小这一特征,提出了利用位点间物理距离,引入甲基化水平相关系数的全局信息,对协方差进行修正的方法cZST.该方法在研究低读数、无差异位点多的BS-seq数据时展示出优于已有方法的功效.通过模拟分析和真实RRBS数据研究,可以看出cZST方法在基因功能区域甲基化差异分析中具有较高灵敏度,并且适合分析测序平均总读数较低的数据.在真实数据处理分析中,对显著性结果进行富集分析,cZST的基因集对应的p值显著功能注释集,拥有与乳腺导管癌更紧密的联系.并且作为一项前瞻性研究,找到了调控Rho基因及Pleckstrin、Dbl基因家族的甲基化与该疾病的密切联系,可为后续设计实验探究乳腺导管癌遗传机理提供帮助. 本方法仍有几个方面值得改进: (1) cZST修正方法局限于协方差矩阵,未能采用如BSmooth方法[32]的策略,利用临近位点甲基化信息对读数小的β值进行修正.(2) 此方法在处理区域内位点间距远、相关系数较小的稀疏数据时,比如模拟实验中c<0.4的情况,表现功效可能不如nZST.因此应根据数据特征决定选用方法.(3) 本方法计算p值时采用置换检验,在基因功能区域协方差、β值确定的情况下,检验的时间取决于置换次数.因此如果多重检验需要对p值作Bonferroni校正时必须保证进行相当数量级的置换次数,检验速度将受到一定影响. 将全部样本甲基化数据β分为病例组βA(affected)与对照组βN(normal).得分向量U的等价表达式为: 从而有 [1] MCCABE M T, DAVIS J N, DAY M L. Regulation of DNA methyltransferase1 by the pRb/E2F1 pathway [J].CancerResearch, 2005,65(9): 3624-3632. [2] VARRIALE A, BERNARDI G. Distribution of DNA methylation, CpGs, and CpG islands in human isochores [J].Genomics, 2010,95(1): 25-28. [3] MA X, WANG Y W, ZHANG M Q,etal. DNA methylation data analysis and its application to cancer research [J].Epigenomics, 2013,5(3): 301-316. [4] KISSELJOVA N P, ZUEVA E S, PEVZNER V S,etal. De novo methylation of selective CpG dinucleotide clusters in transformed cells mediated by an activated N-ras [J].InternationalJournalofOncology, 1998,12: 203-209. [5] JONES P A. Functions of DNA methylation: islands, start sites, gene bodies and beyond [J].NatureReviewsGenetics, 2012,13(7): 484-492. [6] SKINNER M K, GUERRERO-BOSAGNA C. Role of CpG deserts in the epigenetic transgenerational inheritance of differential DNA methylation regions [J].BMCGenomics, 2014,15(1): 692. [7] LI N, YE M, LI Y,etal. Whole genome DNA methylation analysis based on high throughput sequencing technology [J].Methods, 2010,52(3): 203-212. [8] MEISSNER A, GNIRKE A, BELL G W,etal. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis [J].NucleicAcidsResearch, 2005,33(18): 5868-5877. [9] ANIRUDDHA C, RODGER E J, STOCKWELL P A,etal. Technical considerations for reduced representation bisulfite sequencing with multiplexed libraries [J].JournalofBiomedicine&Biotechnology, 2012,2012(4): 741542. [10] WARDEN C D, LEE H, TOMPKINS J D,etal. COHCAP: An integrative genomic pipeline for single-nucleotide resolution DNA methylation analysis [J].NucleicAcidsResearch, 2013,41(11): e117. [11] HEBESTREIT K, DUGAS M, KLEIN H U. Detection of significantly differentially methylated regions in targeted bisulfite sequencing data [J].Bioinformatics, 2013,29(13): 1647-1653. [12] SUN S, YU X. HMM-Fisher: identifying differential methylation using a hidden Markov model and Fisher’s exact test [J].StatisticalApplicationsinGenetics&MolecularBiology, 2016,15(1): 55-67. [13] ROBINSON M D, KAHRAMAN A, LAW C W,etal. Statistical methods for detecting differentially methylated loci and regions [J].FrontiersinGenetics, 2014,5: 324. [14] PAN W, KIM J, ZHANG Y,etal. A powerful and adaptive association test for rare variants [J].Genetics, 2014,197(4): 1081-1095. [15] SCHOOFS T, ROHDE C, HEBESTREIT K,etal. DNA methylation changes are a late event in acute promyelocytic leukemia and coincide with loss of transcription factor binding [J].Blood, 2013,121(1): 178-187. [16] WANG C, SUN L, ZHENG H,etal. Detecting multiple variants associated with disease based on sequencing data of case-parent trios [J].JournalofHumanGenetics, 2016,61(10): 851-860. [17] DOLZHENKO E, SMITH A D. Using beta-binomial regression for high-precision differential methylation analysis in multifactor whole-genome bisulfite sequencing experiments [J].BMCBioinformatics, 2014,15(1): 215. [19] ABBA M C, GONG T, LU Y,etal. A molecular portrait of high-grade ductal carcinomainsitu[J].CancerResearch, 2015,75(18): 3980-3990. [20] SLATTERY M L, LUNDGREEN A, JOHN E M,etal. MAPK genes interact with diet and lifestyle factors to alter risk of breast cancer: The breast cancer health disparities study [J].Nutrition&Cancer, 2015,67(2): 1-13. [21] GRAHAM T R, YACOUB R, TALIAFERROSMITH L,etal. Reciprocal regulation of ZEB1 and AR in triple negative breast cancer cells [J].BreastCancerResearchandTreatment, 2010,123(1): 139-147. [22] SILBERSTEIN G B, VAN H K, STRICKLAND P,etal. Altered expression of the WT1 wilms tumor suppressor gene in human breast cancer [J].ProcNatlAcadSciUSA, 1997,94(15): 8132-8137. [23] CHENG Q, CHANG J T, GERADTS J,etal. Amplification and high-level expression of heat shock protein 90 marks aggressive phenotypes of human epidermal growth factor receptor 2 negative breast cancer [J].BreastCancerResearch, 2012,14(2): R62. [24] PEDERSON H J, PADIA S A, MAY M,etal. Managing patients at genetic risk of breast cancer [J].CleveClinJMed, 2016,83(3): 199-206. [25] YONG W, LEUNG F C C. An evaluation of new criteria for CpG islands in the human genome as gene markers [J].Bioinformatics, 2004,20(7): 1170-1177. [26] HUANG D W, SHERMAN B T, LEMPICKI R A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources [J].NatureProtocol, 2009,4(1): 44-57. [27] FRIXEN U H, BEHRENS J, SACHS M,etal. E-cadherin-mediated cell-cell adhesion prevents invasiveness of human carcinoma cells [J].JournalofCellBiology, 1991,113(1): 173-185. [28] HU X C, LOO W T, CHOW L W. E-cadherin promoter methylation can regulate its expression in invasive ductal breast cancer tissue in Chinese woman [J].LifeSciences, 2002,71(12): 1397-1404. [30] HALON A, DONIZY P, SUROWIAK P,etal. ERM/Rho protein expression in ductal breast cancer: A15 year follow-up [J].CellularOncology, 2013,36(3): 181-190. [31] MURAKAMI E, NAKANISHI Y, HIROTANI Y,etal. Roles of ras homolog a in invasive ductal breast carcinoma [J].ActaHistochemicaEtCytochemica, 2016,49(5): 131-140. [32] HANSEN K D, IRIZARRY R A, ZHIJIN W. Removing technical variability in RNA-seq data using conditional quantile normalization [J].Biostatistics, 2012,13(2): 204-216. AStatisticalMethodforDetectingDifferentiallyMethylatedGeneFunctionalRegionsBasedonCase-ControlStudies MAXinyu,WANGChan,HUYueqing (DepartmentofBiostatisticsandComputationalBiology,SchoolofLifeSciences,FudanUniversity,Shanghai200433,China) DNA methylation is an important epigenetic modification that participates in the regulation of gene expression and other biological activities. In case-control studies, searching for differentially methylated gene functional regions associated with disease can provide clues of disease-related gene list for follow-up studies. Nowadays existing differential methylation detection methods are lack of a uniform standard for defining differentially methylated regions. Also test results can be across multiple genes or multiple functional regions, bringing difficulty for biological interpretation. In this paper, a statistical approach, cZST, is proposed based on Z-score test. It is able to detect differences of methylation levels in each gene functional region. This method uses physical distances between sites and the global information of methylation levels in each chromosome to modify the covariance matrix. The simulation results show that cZST is superior to other methods in BS-seq data of a small sample size or a low reading depth. In the real data analysis of a breast ductal cancer study, 661 significant functional regions and 81 significant single loci are recognized. Gene annotation analysis of results demonstrates the effectiveness of cZST. Also we find that in regulator genes of Rho,Dbl and Pleckstrin gene families, DNA methylation level differences are associated with breast ductal carcinoma affection. differential methylation detection; BS-seq; functional enrichment analysis 0427-7104(2017)06-0701-11 2017-04-05 国家自然科学基金(11571082,11171075) 马欣宇(1992—),男,硕士研究生;胡跃清,男,教授,通信联系人,E-mail: yuehu@fudan.edu.cn Q348 A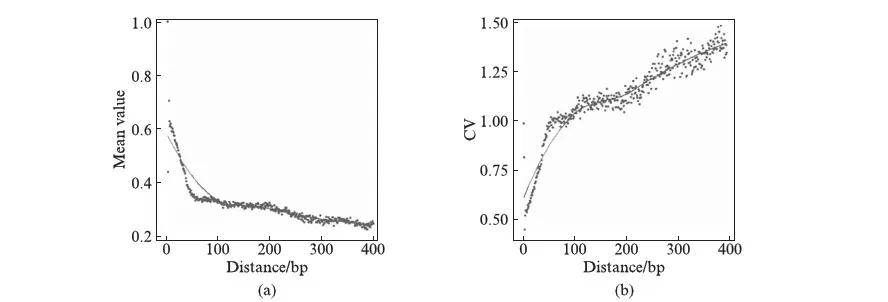
2 随机模拟研究

3 真实数据分析


4 总结与展望
5 附 录
猜你喜欢
植物保护(2019年2期)2019-07-23 08:40:58
中国公路(2017年19期)2018-01-23 03:06:36
中国公路(2017年15期)2017-10-16 01:32:04
中国公路(2017年9期)2017-07-25 13:26:38
中国公路(2017年7期)2017-07-24 13:56:40
湖北农业科学(2016年24期)2017-03-18 00:35:21
吉林农业(2016年12期)2017-01-06 19:24:54
自动化学报(2016年8期)2016-04-16 03:38:55
无线电通信技术(2015年3期)2015-12-23 11:37:00
中国科学技术大学学报(2013年8期)2013-03-11 20:18:37
