
果糖1,6二磷酸酶缺乏症的遗传学诊断及突变位点分析
2018-01-03 07:27赵银霞于辛酉
临床儿科杂志 2017年12期
赵银霞 梁 娟 刘 静 陆 彪 于辛酉
宁夏医科大学总医院1.儿科,2.医学实验中心(宁夏银川 750004)
果糖1,6二磷酸酶缺乏症的遗传学诊断及突变位点分析
赵银霞1梁 娟1刘 静1陆 彪1于辛酉2
宁夏医科大学总医院1.儿科,2.医学实验中心(宁夏银川 750004)
目的 探讨果糖1,6二磷酸酶缺乏症的遗传学诊断及致病基因的突变位点分析。方法 回顾1例果糖1,6二磷酸酶缺乏症患儿的临床资料及相关基因的panel筛选结果。结果 2岁女孩,反复感染后出现恶心、呕吐、精神差及嗜睡,伴有间断抽搐。血生化检查示低血糖症、酸中毒;FBP1基因存在错义变异c.355G>A,p.Asp119Asn(纯合),父母均携带该位点变异(杂合)。结论 对于反复感染后出现发作性低血糖、酸中毒的患儿,需考虑果糖1,6二磷酸酶缺乏症的可能。
果糖1,6-二磷酸酶缺乏; FBP1基因; 低血糖症; 酸中毒
果糖1,6二磷酸酶(fructose-1, 6-bisphosphatase,FBPase)缺乏症(OMIM 229700)是一种常染色体隐性遗传病,致病基因为FBP1(OMIM 611570),定位于9q22.2~q22.3,全长约31 kb,含8个外显子[1]。发病率在荷兰与法国人群中分别为1/350 000、<1/90 000[2,3]。该基因的突变可导致肝脏的果糖1,6二磷酸酶缺乏或活性低下,造成1,6二磷酸果糖不能转化为6-磷酸果糖,影响糖异生的过程。反复感染后发作性的低血糖、酸中毒及酮症是本病的主要临床表现。尽管该病在新生儿期为致死性疾病,然而及早诊断和有效治疗仍可获得较好预后[4,5]。现报1例经过相关基因检测得到确诊的果糖1,6二磷酸酶缺乏症患儿,并结合文献进行分析,提高对本病的认识水平。
1 临床资料
患儿,女,2岁,因“发热、意识不清4天,呕吐3天,抽搐1次”收入宁夏医科大学总医院。患儿每隔4~6个月由于感染后间断出现恶心、呕吐及嗜睡,唤醒后语言能对答。患儿G1P1,足月顺产,父母体健,非近亲婚配,两个妹妹体健,无特殊疾病家族史。入院体检:神志不清,疼痛刺激时有肢体活动;无自主睁眼,双侧瞳孔等大等圆,光反应存在;双眼窝凹陷,口唇干燥,皮肤弹性可;呼吸深大,双肺呼吸音粗,未闻及啰音;心音略低钝,节律齐,心前区未闻及杂音;腹软,肝脏肋下2 cm,质尚软,脾脏肋下未及,肠鸣音正常;颈软,生理反射未引出,克氏征、巴氏征阴性;Glasgow评分6分(无自发睁眼1分、因局部疼痛而动4分,无语言反应1分)。实验室检查:颅脑CT平扫未见异常;血气分析示pH 值7.11,二氧化碳分压10.00 mmHg,氧分压113.00 mmHg,碱剩余-25.6 mmol/L;血生化常规示总蛋白76.6 g/L,白蛋白46.9 g/L,丙氨酸氨基转移酶83.5 U/L,天冬氨酸氨基转移酶117.8 U/L,尿素23.90 mmol/L,肌酐142.7 μmol/L,钾7.32 mmol/L,钠130.6 mmol/L,肌酸激酶同工酶180.0 U/L,葡糖糖6.6 mmol/L,镁1.48 mmol/L,超敏C反应蛋白2.46 mg/L;凝血全套示 凝血酶原时间(PT) 12.1 s,活化部分凝血活酶时间(APTT)25.0 s,纤维蛋白原(FIB)1.122 g/L。脑脊液生化检查示总蛋白0.370 g/L,葡萄糖3.5 mmol/L;脑脊液常规检查示细胞总数7/mm3,白细胞计数0/mm3;脑脊液涂片(-);脑脊液细胞学检查示:红细胞计数0/mm3,白细胞计数5/mm3。腹部B超未见异常。行血糖检测为低血糖,经治疗后7~10天患儿血糖水平恢复正常。期间患儿曾行血、尿遗传代谢病筛查,未能明确诊断。
经医学伦理委员会审核并,家属签署知情同意书后,采集患儿及父母的外周血2~3 mL,进行基因检测。以 Illumina NextSeq 500(© 2017 Illumina, Inc)为主的平台进行测序。测序数据经NextGene软件(Softgenetics, State College, PA)进行匹配分析,利用Ingenuity在线软件系统(Ingenuity☒ Pathway Analysis IPA☒,© QIAGEN 2017)进行突变位点分析。采用遗传性疾病panel(2 742个基因)进行遗传代谢性疾病相关基因的检测,检测的基因覆盖氨基酸代谢病、有机酸代谢病、糖代谢障碍、溶酶体贮积症等遗传性代谢病,包括半乳糖血症、戈谢病、酪氨酸血症、果糖不耐症、果糖1,6二磷酸缺乏症、糖原累积病、间质性肾炎等等。对于测序的阳性结果(疑似致病位点和致病性位点),采用Primer Premier 3.0软件(©1994—2017 PREMIER Biosoft)进行引物设计、利用ABI3130平台(©2016 Thermo Fisher Scientific Inc.)进行Sanger测序验证及家系验证。
在患儿的外周血中检测到FBP1基因的3号外显子中存在纯合突变c.355G>A(图1),由于该突变导致编码的氨基酸由天冬氨酸(Asp)变为天冬酰胺(Asn),其他基因未发现病理性变异。家系验证结果提示:患儿父母均携带该位点突变(图2,3)。

图1 FBP1基因检测高通量测序结果
患儿在急性发作期积极纠正低血糖,静脉给予输入葡萄糖0.5 g/(kg·h),合并代谢性酸中毒给予碳酸氢钠纠正酸中毒,伴有脱水症状及时补液支持治疗。日间少量多次喂食碳水化合物的食物,夜间使用鼻饲点滴葡萄糖,10 mg/(kg·min)维持,使血糖维持在4~5 mmol/L。维持治疗期间避免饥饿,在合并有发热、感染及应激的情况下,增加喂养次数及食用生玉米淀粉,必要时给予鼻胃管持续喂养。经上述治疗后,7~10天患儿精神及活动恢复正常。目前患儿体格及智力发育水平正常。
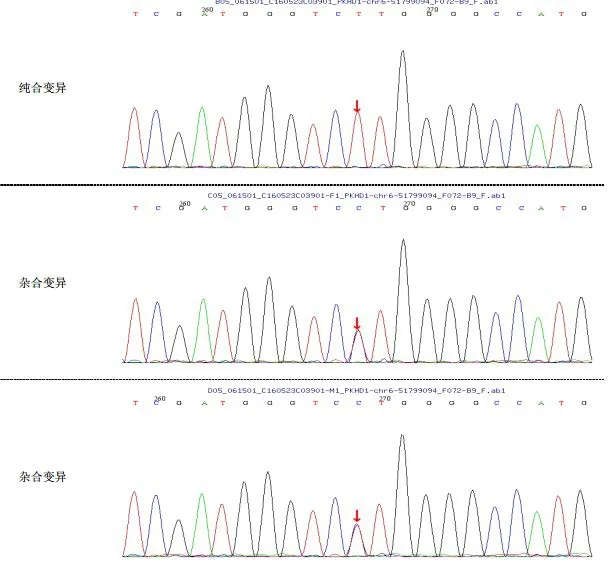
图2 FBP1基因序列(Sanger测序)

图3 FBP1基因家系分析结果
2 讨论
果糖1,6二磷酸酶缺乏症是一种罕见的常染色体隐性遗传病,早于1970年Baker报道[6],由于FBP1基因突变导致肝脏果糖1,6二磷酸酶缺乏或活性低下,引起糖异生障碍。葡萄糖异生的过程中,果糖1,6二磷酸酶是糖异生途径中的关键催化酶,由于该酶的缺陷导致1,6二磷酸果糖不能向6-磷酸果糖转化,引起糖异生底物转化为葡糖糖障碍,导致低血糖、酸中毒等。文献报道,果糖1,6二磷酸酶缺乏症往往有低血糖症的临床表现,面色苍白、头痛、头晕、四肢凉、发绀及呼吸暂停,同时可有脑功能紊乱,无力、嗜睡、精神改变、认知障碍、抽搐及昏迷,反复的低血糖症可引起脑细胞能量不足,代谢衰竭,婴幼儿是脑发育关键时期,严重的低血糖可出现脑损伤,导致精神运动发育迟滞甚至猝死[7]。
本例患儿自生后2岁开始,反复出现发作性的拒食、嗜睡及惊厥。第一次入院前由于静滴葡糖糖,入院后查患儿血糖正常。患儿每4~6个月由于感染后间断出现恶心、呕吐、嗜睡及抽搐,未给予葡糖糖治疗的血生化检查明确患儿存在低血糖、酸中毒。反复出现发作性的低血糖、酸中毒考虑存在能量代谢障碍性疾病,此患儿的相关检查不支持内分泌性疾病,遗传代谢性疾病不能除外。通过遗传代谢性基因panel检测到FBP1基因突变,临床表现与基因型均符合FBPase缺乏症的诊断。
有研究者在果糖1,6二磷酸酶患儿中检测到FBP1基因的突变,其中在13例患者中检测到10例含FBP1基因的纯合突变或复合杂合突变,突变类型为插入突变(960insG),提示该突变是FBP1基因较常见的突变类型[5,8]。该变异也是高加索人及中国人的常见变异[3,5, 8,9]。
本例患儿检测到FBP1基因突变,突变点位于Chr9∶94617839 (GRCh38),位于3号外显子,NM_000507.3 c.355G>A(p.Asp119Asn)。该突变位点暂未见公开文献报道,截止2017年2月,ExAC数据库(http∶//exac.broadinstitute.org)显示,FBP1基因的该处突变频率为0.001 65%(2/121 018),远远低于1/10 000,突变位点所在人群分别为东亚和欧洲裔。在Clinvar数据库(https∶//www.ncbi.nlm.nih.gov/clinvar)可查询到1例结果:GenxDx公司于2014年提交的检测结果为(FBP1)∶c.355G>A (p.Asp119Asn),与本例相同。FBP1基因c.355G>A的错义突变,使得编码的氨基酸由天冬氨酸(Asp)变为天冬酰胺(Asn),该突变类型是一种半保守的氨基酸替换,可影响蛋白质的二级结构。经Alamut软件预测分析:其可能会影响蛋白结构域的功能。同时该变异在家系验证过程中,也表现出与疾病的共分离现象,根据ACMG 2016年发布的基因变异解读标准及指导原则[10]应可判断该位点为“致病性位点”。关于该突变位点如何引起该病的发生需进行大量的深入研究。
本例患儿的家系分析结果提示,该突变位点来源于父母,父母分别为携带者。该结果符合常染色体隐性遗传的遗传方式(autosomal recessive,AR)。AR的特征为致病基因位于常染色体,男女发病机会均等,与性别无关;多为散发病例,同胞中可有多人患病;双亲一般不患病,是致病基因的携带者;再发风险为1/4,表型正常的子女中有2/3的概率是携带者[11]。故患儿父母再次生育时仍存在1/4的发病风险,可选择植入前遗传学诊断或产前诊断。
该病在急性发作期应积极纠正低血糖,抑制低血糖所继发的各种代谢紊乱。可给予静脉输入葡萄糖,静滴碳酸氢钠、补液等治疗,使血糖浓度维持在4~5 mmol/L,应避免饥饿。在合并有发热、感染及应激的情况下,应增加喂养次数及食用生玉米淀粉,血糖不能维持必要时给予鼻胃管持续喂养。给予上述治疗后,患儿大多于发病7~10天左右患儿精神及活动可恢复正常。
持续反复发作性的低血糖,同时伴有酸中毒、酮症患儿,行血尿串联质谱不能明确诊断时要考虑1,6二磷酸酶缺乏症的可能,行FBP 1基因检测可早期诊断,及时治疗纠正低血糖可避免猝死及永久性的脑损伤,提高患儿的生活质量。同时需进行相应的遗传咨询并告知再发风险,为再次生育提供合理建议。
[1] el-Maghrabi MR, Lange AJ, Jiang W, et al. Human fructose-1,6-bisphosphatase gene (FBP1): exon-intron organization,localization to chromosome bands 9q22.2-q22.3, and mutation screening in subjects with fructose-1,6-bisphosphatase def i ciency [J]. Genomics, 1995, 27(3): 520-525.
[2] Krishnamurthy V, Eschrich K, Boney A, et al. Three successful pregnancies throμgh dietary management of fructose-1,6-bisphosphatase deficiency [J]. J Inherit Metab Dis, 2007, 30(5): 819-819.
[3] Lebigot E, Brassier A, Zater M, et al. Fructose 1,6-bisphosphatase def i ciency: clinical, biochemical and genetic features in French patients [J]. J Inherit Metab Dis, 2015,38(5): 881-887.
[4] Matsuura T, Chinen Y, Arashiro R, et al. Two newly identif i ed genomic mutations in a Japanese female patient with fructose-1,6-bisphosphatase (FBPase) deficiency [J]. Mol Genet Metab, 2002, 76(3): 207-210.
[5] Kikawa Y, Inuzuka M, Jin BY, et al. Identification of genetic mutations in japanese patients with Fructose-1,6-bisphosphatase deficiency [J]. Am J Hum Genet, 1997,61(4): 852-861.
[6] Baker L, Winegrad AI. Fasting hypoglycaemia and metabolic acidosis associated with deficiency of hepatic fructose-1,6-diphosphatase activity [J]. Lancet, 1970, 2(7662): 13-6.
[7] Kikawa Y, Inuzuka M, Jin BY, et al. Identification of a genetic mutation in a family with fructose-1,6-bisphosphatase deficiency [J]. Biochem Bioph Res Co, 1995, 210(3): 797-804.
[8] Herzog B, Morris AAM, Saunders C, et al. Mutation spectrum in patients with fructose-1,6-bisphosphatase def i ciency [J]. J Inherit Metab Dis, 2001, 24(1): 87.
[9] Xu K, Liu XQ, Zhang CY, et al. Genetic diagnosis of fructose-1, 6-bisphosphatase deficiency: a case report [J].Beijing Da Xue Xue Bao, 2014, 46(5):681-685.
[10] Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [J]. Genet Med, 2015, 17(5):405-424.
[11] 邬玲仟, 张学. 医学遗传学[M]. 北京: 人民卫生出版社,2016.
Genetic diagnosis and mutation site analysis of fructose 1, 6 diphosphatase def i ciency
ZHAO Yinxia1, LIANG Juan1,LIU Jing1, LU Biao1, YU Xinyou2
(1.Department of Pediatrics, 2.Department of Medical Experimental Center, General Hospital of Ningxia Medical University, Yinchuan 75004, Ningxia, China)
Objectives To explore the genetic diagnosis of fructose 1,6 diphosphatase def i ciency and analysis of mutation sites of its pathogenic genes. Methods The clinical data and the related results of gene panel screening in one child with fructose 1, 6 diphosphatase (FBPase) def i ciency were retrospectively reviewed. Results The 2-year-old girl suffered repeated infection, nausea, vomiting, mental illness, and drowsiness, accompanied by intermittent convulsions. Blood biochemical tests sμggested hypoglycemia and acidosis. The FBP1 gene had a missense mutation, c.355G>A, p.Asp119Asn (isozygoty). Both her parents carried the locus variation (heterozygous). Conclusions Fructose 1, 6 diphosphatase def i ciency should be considered when child with hypoglycemia after repeated infection, acidosis, and ketosis.
fructose 1,6 diphosphatase def i ciency; FBP1 gene; hypoglycemia; acidosis
doi∶10.3969/j.issn.1000-3606.2017.12.001
宁夏自然科学基金(No.NZ17175)
于辛酉 电子信箱:yuxinyou@nxmu.edu.cn
2017-06-06)
邹 强)
猜你喜欢
今日畜牧兽医(2022年10期)2022-12-23
今日农业(2021年16期)2021-11-26
湖南饲料(2021年4期)2021-10-13
现代畜牧科技(2021年4期)2021-07-21
现代畜牧科技(2021年6期)2021-07-16
现代畜牧科技(2021年6期)2021-07-16
猪业科学(2018年5期)2018-07-17
中国组织化学与细胞化学杂志(2017年1期)2017-06-15
中国民族医药杂志(2016年2期)2016-05-14
医学研究杂志(2015年8期)2015-06-22
