
熔盐电解法乏燃料干法后处理技术研究进展
2017-12-20 09:43任一鸣
核化学与放射化学 2017年6期
唐 浩,任一鸣,邵 浪,钟 毅,高 瑞
中国工程物理研究院 材料研究所,四川 江油 621907
熔盐电解法乏燃料干法后处理技术研究进展
唐 浩,任一鸣,邵 浪,钟 毅,高 瑞
中国工程物理研究院 材料研究所,四川 江油 621907
熔盐电解法是目前最有前途的干法后处理技术,适合于处理氧化物和金属等不同类型乏燃料。熔盐电解法主要包括四个核心流程,即首端处理、电解还原、电解精炼和提取以及废物处理。本文以国际上最新的研究进展为蓝本,综述熔盐电解法乏燃料后处理技术的基本流程以及待解决的关键问题。
乏燃料;干法后处理;核废物;熔盐电解;锕系元素
核能是可被人类大规模利用的清洁能源,用核电替代化石燃料发电能够大规模减少温室气体的排放,对于环境污染日益严峻的我国,核能有着不可替代的吸引力。到2020年,我国核电装机容量和在建容量将分别达到5 800万千瓦和3 000万千瓦以上。核能的可持续发展必须解决铀资源充分利用和核废物最少化两大主要问题。只有采取核燃料闭式循环方式,对乏燃料进行后处理,才能实现上述目标[1]。因此,开展乏燃料的后处理工艺路线及关键技术研究,对于我国核能的可持续发展具有重要意义。
乏燃料是经过反应堆辐照后取出的核燃料,其主要成分是铀和钚等核裂变材料,有害的裂变产物只占4%左右,仍然具有极大的经济价值,而不能简单的将其看作是核废料[2]。通过对乏燃料进行化学处理,实现锕系元素和裂变产物的分离。将回收的铀和钚重新制造成核燃料供给快堆燃烧发电,同时将分离出的次锕系元素在快堆或加速器中焚烧,则铀资源的利用率可提高50~60倍,且高放废物的体积和长期毒性将降低1~2个数量级[3-4]。
随着我国快堆技术的迅速发展,研发并掌握先进乏燃料后处理技术已迫在眉睫。熔盐电解法乏燃料后处理技术在处理不同类型的快堆乏燃料领域具有广阔的应用前景,发展相关技术对我国实现快堆核燃料闭式循环具有重要意义。近年来,我国从顶层设计入手,规划熔盐电解法乏燃料干法后处理技术的发展,在《能源技术革命创新行动计划(2016—2030年)》中,明确提出重点研发与攻关适用于快堆等的先进燃料循环的干法后处理技术,到2030年,“提出干法后处理技术的优选路线,建成具备公斤级熔盐电解分离铀、钚的实验装置”。为了进一步推动我国在熔盐电解干法后处理领域的发展,本文将综述熔盐电解法乏燃料后处理技术的最新研究进展。
1 熔盐电解法概述
由于材料和工程技术的不断进步,20世纪中后期国际上掀起了熔盐电解(也称作高温冶金或高温化学)乏燃料干法后处理技术的研究热潮[5-8]。在一体化快堆计划的推动下,20世纪80年代美国提出采用氯化物熔盐电解精炼流程(electrorefining)来回收金属乏燃料中铀和超铀元素[9-10]。在1996—1999年期间,美国采用熔盐电解精炼法成功地处理了来自EBR-Ⅱ型快堆的0.4 t驱动燃料和0.7 t增殖燃料,回收了金属铀,并且只产生了少量的陶瓷和金属核废料,利于进行长期地质贮存,证明了熔盐电解精炼法适合处理金属乏燃料[11]。紧接着,美国又开展了熔盐电解提取流程(electrowinning)工程化的研发工作,用于回收钚和次锕系元素[12]。日本、韩国和欧洲在美国研究工作的基础上,积极自主研发熔盐电解精炼和提取流程,并验证了可行性[13-15]。但是,熔盐电解精炼法无法直接处理目前主流的氧化物乏燃料,极大地限制了其应用范围。近年来,美国、日本和韩国等[16-18]积极研发熔盐电解还原技术(electrochemical reduction),能够将氧化物乏燃料转化为金属乏燃料,再结合熔盐电解精炼技术,可用来处理轻水堆和快堆的氧化物乏燃料,扩大了熔盐电解法的应用范围,完善了核燃料闭式循环体系。此外,从20世纪60年代,俄罗斯开始研发能够用于处理快堆MOX(mixed oxide fuel)燃料的干法后处理技术,并采用经过研究堆辐照后的MOX乏燃料进行了半工业化流程验证[19]。其核心流程也是在熔盐电解池中完成的,产品是UO2、PuO2和MOX,可直接用于制造MOX燃料。
总体来说,熔盐电解法后处理技术具有以下特点:(1) 具有处理不同类型和短冷却期乏燃料的能力;(2) 流程简单,设备结构紧凑,装置体积小,可紧邻核电站建设,实现闭式循环;(3) 采用高温无机盐作为溶剂体系,具有耐辐照、低临界风险的特点;(4) 不制造纯钚,防止核扩散;(5) 回收超铀元素,产生少量固体核废料,易于永久地质埋藏。上述优点使得熔盐电解法成为当前研究最活跃的干法后处理技术。目前,国际上积极研究开发熔盐电解法后处理技术的主要科研机构有美国阿贡国家实验室(ANL)和爱达荷国家实验室(INL)、韩国原子能研究院(KAERI)、日本电力公司中央研究所(CRIEPI)、欧盟超铀元素联合研究中心(JRC-ITU)、印度甘地原子能研究中心(IGCAR)、俄罗斯原子反应堆研究院(RIAR),我国的中国原子能科学研究院(CIAE)、中国科学院高能物理研究所(IHEP)、哈尔滨工程大学(HEU)以及中国工程物理研究院(CAEP)等。
2 熔盐电解法核心流程
由于熔盐电解法涉及的技术和学科种类极其繁多,下面将主要介绍以美国和韩国等国大力发展的熔盐电解法的核心技术流程,即首端处理、电解还原、电解精炼和提取以及废物处理四个技术流程。至于其它相关技术以及俄罗斯研发的熔盐电解干法后处理技术请参阅其它相关文献资料[2,8,19]。
图1是熔盐电解法后处理氧化物乏燃料的主要流程示意图。首端处理作为乏燃料后处理的第一步,其首要目的是将氧化物乏燃料与锆包壳分离,主要包括切割(chopping)、去包壳(decladding)、氧化挥发(voloxidation)和废气处理(off-gas treatment)等四个步骤,其核心技术为氧化挥发流程。熔盐电解技术主要包括电解还原、电解精炼和电解提取三个流程,其任务是回收锕系元素。废物处理流程(wastes treatment)的主要研究对象是高放熔盐,其目的是尽可能减少高放废物的数量并将高放废物转化为适合永久地质埋藏的稳定形态。
2.1 首端处理
在设计首端处理流程时,遵循两个目标:(1) 操作简单,费用低廉;(2) 最大程度地将乏燃料与包壳分离。首端处理流程需要对乏燃料进行精密操作,且这些操作全部由远程控制的机械手完成。因此能否设计出一整套控制简单、操作精确的机械手也是核心任务。KAERI设计的机械手对位置定位的误差控制在0.5 mm以内,满足了热室远程控制的要求[18]。

图1 典型熔盐电解法后处理氧化物乏燃料的流程图Fig.1 Typical flow chart for pyroprocessing of spent oxide fuel
INL和KAERI合作开发的首端处理流程主要包括四个步骤:切割、去包壳、氧化挥发和废气处理[20-21]。通过首端处理,不仅实现了乏燃料与包壳材料的分离,而且能够去除挥发性裂变产物,包括气体废物以及钼、锝、铑、钌和铯等,同时可以制造出大小合适的氧化物颗粒供给熔盐电解还原流程[22-24]。
切割流程是乏燃料后处理的第一步。首先通过远程控制的机械手拆卸核燃料元件并取出燃料棒。然后剪切掉燃料棒的上端装置(内含裂变气体产物),并利用激光切割机将燃料棒切割成大小合适的小元件[18]。
去包壳是首端处理的重要步骤。近年来,INL和KAERI采用氧化去包壳法分离乏燃料和包壳材料[24-25]。切割好的燃料棒在有空气或氧气的条件下加热至500~700 ℃进行氧化反应。在此过程中,二氧化铀被氧化成八氧化三铀,其它锕系氧化物也被氧化成更稳定的氧化物,贵金属也继续被氧化成高化合价氧化物,但碱金属、碱土金属和稀土氧化物没有变化。氧化过程中,乏燃料的体积将增大约30%,但由于包壳的裹覆,使得元件内部压力增大,最终包壳由于压力增大而膨胀裂开。在此过程中,氧化物乏燃料被粉碎成细小的粉末,易于从包壳中分离,分离效率超过99%。但是当加热温度达到800 ℃时,氧气会与包壳材料发生反应,反而降低了分离效率。需要指出的是,在乏燃料粉末化过程中,会有挥发性裂变产物逸出(主要是氚),需要有效地收集。
氧化挥发和废气净化流程的目的是去除乏燃料中易挥发的裂变产物,以便简化下游电化学流程。INL和KAERI重点研究了在不同温度和压力下,乏燃料与包壳材料的分离效率和挥发性裂变产物净化效率[21,25-26]。为了防止坩埚材质吸附挥发性裂变产物,INL使用铬镍铁合金坩埚;并且为了充分氧化乏燃料,燃料棒被竖直摆放在坩埚中。加热温度为300~1 400 ℃,吸收挥发产物装置有浸银沸石过滤器(silver impregnated zeolite filter)、氧化钙过滤器(calcium oxide filter)和煤飞灰过滤器(fly ash filter)等过滤装置。首先,在空气或氧气的气氛下,以10 min/℃的速率将乏燃料加热至500 ℃,然后保温3 h。在氧化过程中,氚会全部逸出,还有少量的碳、氪、氙和碘。氚与氧气反应,生成水蒸气。然后,将体系抽真空,再以10 min/℃的速率加热至1 200 ℃。此时,裂变气体由下至上依次经过上述的过滤装置,分别用来捕获铯和碲,吸收钌、锝、钼和碳以及吸附碘等。当气体走完废气处理装置后,会被引入分子筛来吸收重水,然后引进银-沸石装置来吸收氙,最后是氢-沸石装置吸收氪。KAERI利用模拟乏燃料实施高温氧化挥发法和废气净化流程得到的挥发性裂变产物的收集效率[23]列入表1。
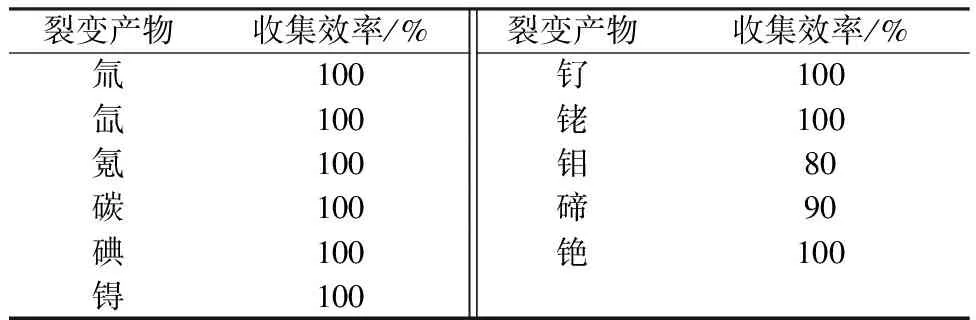
表1 裂变产物的收集效率[23]Table 1 Collection efficiency of fission products[23]
2.2 熔盐电解还原
在21世纪初,受到熔盐电解还原二氧化钛[27]的启发,ANL[28]将熔盐电脱氧技术应用到后处理氧化物乏燃料领域。针对熔盐电脱氧效率较低的缺点,ANL将熔盐电脱氧技术与锂热还原技术相结合,开发了熔盐电解还原氧化物乏燃料技术[29]。随后,KAERI[30]和CRIEPI[31]在美国流程基础上,自主开展了大量的研发工作,提出了改进流程。目前,熔盐电解还原流程的研究内容和方向主要集中在锕系氧化物还原机理和效率、乏燃料各组分的分布、阳极材料、多孔惰性材料和新熔盐体系。
熔盐电解还原技术通常以氧化物乏燃料作为阴极,铂电极作为阳极,LiCl-Li2O(通常是Li2O(w=1%))熔盐体系为电解质,操作温度为650 ℃,多采用控制电流方式电解,如图2所示。通电后,一方面,阴极氧化物中氧元素在电子作用下离子化变为氧离子溶解到熔盐中,经过熔盐体系迁移到阳极氧化为氧气从电解池中逸出,在阴极留下金属。另一方面,电解质中氧化锂也被同时电解分离,在阴极产生金属锂,阳极产生氧气;原位生成的金属锂可以迅速地将阴极氧化物还原成金属,同时自身又转化为氧化锂,实现了氧化锂的自循环。

图2 熔盐电解还原流程示意图Fig.2 Electrolytic reduction process
熔盐电解还原过程机理研究[32-33]表明,氧离子氧化为氧气的阳极反应是速率控制步骤。电解还原流程中O2-迁移过程包括:O2-从阴极样品中扩散到外表面,O2-溶解到熔盐中,O2-向阳极迁移等。因此,O2-在熔盐体系中的溶解度和扩散速率对电解还原流程的速率和效率有重要影响。理论计算和实验研究表明,电解质中Li2O初始质量分数合适的范围为0.5%~3.0%。在电解还原初期,电脱氧过程起主导作用,后期以锂热还原为主,其原因是当表面覆盖上还原金属后,氧元素很难从阴极内部扩散到外部、进而离子化溶解到熔盐中,而熔盐和金属锂可以渗透到阴极内部,进行还原反应[34-36]。此外,根据IGCAR研究,纯CaCl2熔盐不适合作为电解质,因为高达800 ℃的操作温度会导致被还原的金属铀在氧化铀表面生成致密的金属层,隔绝了氧化铀与电解质的接触,阻止了氧离子进一步扩散到熔盐中,导致电解还原反应无法继续进行[37-38]。
在电解还原过程中,氧化物原料的结构,阴极篮的材料和结构,阴极和阳极电极面积比例以及距离,阴极UO2/U3O8的形态,阳极的材料和结构等[39-43]都对电解还原效率和速率有重要影响。特别是氧化物原料应满足以下条件:(1)具备多孔结构,利于快速深度还原;(2)具备稳定的固态结构,利于安全稳定操作,同时还原过程不会掉落进入熔盐中,减少锕系元素损耗。因此,氧化挥发流程得到的粉末状氧化物需要进一步加工,例如烧结成多孔小球,这样更利于简便操作。
目前,INL已进行了真实乏燃料实验室级别的电解还原试验,包括轻水堆BR-3[44]和快堆MOX[45]乏燃料。在BR-3燃料试验中,碱金属和碱土金属以及稀土元素铕溶解到了熔盐中,稀土以及贵金属元素和锕系元素仍保留在阴极中。分别有99.7%(w)的二氧化铀和97.8%的二氧化钚被还原成金属铀和钚,其它锕系元素的还原率也超过了90%。同时,稀土元素的还原率低于80%,贵金属主要以氧化物形式存在,少量的被还原后溶解在生成的锕系金属相中。但是,电解还原试验的电流效率较低,最高批次的电流效率也只有45%。在快堆MOX乏燃料试验中,除了碱金属和碱土金属溶解到了熔盐中,碲和碘也在熔盐中积累,稀土以及贵金属和锕系元素仍保留在阴极电极中。但是,锕系元素的还原率明显低于轻水堆乏燃料,二氧化铀的还原率最高为88%,而二氧化钚只有56%。稀土氧化物几乎未被还原,贵金属部分被还原。在上述试验中,阳极铂电极遭受严重腐蚀,约损耗了20%的铂电极。为了能够更好地控制和改进电解还原流程,INL[46-47]正在努力建立电解还原过程的动力学模型和扩散模型。韩国已经完成20公斤级别的熔盐电解还原模拟氧化物乏燃料试验。结果表明,氧化铀的还原率达到99.6%,而稀土氧化物还原率只有50%左右,贵金属还原率更低[48-49]。日本也开展了熔盐电解法处理百克级二氧化铀以及模拟氧化物乏燃料的研究[31,50]。结果表明,金属铀的还原率高达99.2%,电流效率高于62%;模拟乏燃料中锶全部溶解到熔盐中,稀土大部分以氧化物形式保留在阴极产物中,少部分锆元素被还原为金属态后与其它金属形成合金产物。
电解还原结束后,阴极吊篮中大约有相当于金属产物质量20%的残留盐。为了减轻残留盐对下游电解精炼过程的影响,KAERI提出用真空蒸馏法分离阴极电极中残留的熔盐。试验结果表明,99%(质量分数)以上的残留熔盐得到分离,且蒸馏后的熔盐接近100%得到回收[18,51]。
2.3 熔盐电解精炼和提取
熔盐电解精炼和提取流程的核心任务是分离锕系元素和裂变产物,分为两个步骤来完成,如图3所示。第一步是实现铀和裂变产物的分离,获得纯的金属铀;第二步是实现超铀元素和裂变产物的分离,获得铀、钚以及次锕系元素的合金。
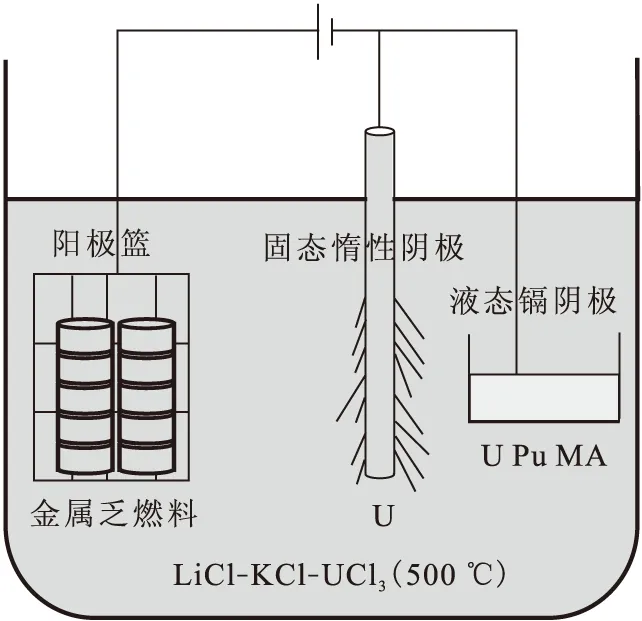
图3 熔盐电解精炼和提取流程示意图Fig.3 Electrorefining and electrowinning processes
电解精炼铀的目的是得到纯的金属铀[52-54]。将金属乏燃料从反应堆中取出后用切割机切成小块燃料,装进不锈钢吊篮中作为阳极或者直接采用电解还原流程阴极得到的还原金属作为阳极,阴极是圆柱形低碳钢,电解质为LiCl-KCl共晶盐,且含有预先溶解的约6%~10%(质量分数)的UCl3,电解温度为500 ℃。一旦将乏燃料浸入熔盐中,在阳极上,钠和裂变产物中的活泼金属(碱金属、碱土金属、稀土金属、超铀金属)会与UCl3发生置换反应,生成氯化物溶解到熔盐中。通电电解后,通过控制电流和电位的大小,使得金属铀从阳极溶解到熔盐中,通过熔盐的传输,在阴极上沉积。理想状态下,锆和贵金属无法进行电化学溶解变成离子态,保留在阳极;活泼金属不能在阴极被还原,在熔盐中不断累积;氯化铀在熔盐中的含量保持不变。最终,在固体阴极上得到纯铀,实现铀和裂变产物的分离。电解完成后,取出阴极和阳极,换上新的阴极和阳极,可以继续电解。
在电解精炼流程中,固态阴极上得到的金属铀产品呈树枝状,金属铀枝晶会沿着某一个方向一直生长,从而造成电解池短路。此外,树枝状产物与固态电极结合不紧密,会从固态阴极上脱落,沉到电解池底部,降低铀的回收率和电流效率[55]。为了防止铀枝晶不断长大,ANL和INL设计了特殊结构的电解装置,在固态阴极上安装了刮刀,可以持续的将阴极产物中较大枝晶从固态阴极上机械剥离到液态镉层或收集篮中[56]。此外,搅拌熔盐是提高电流效率和沉积物质量的好方法。根据INL实验结果,通过阳极搅拌,即电解时,阳极装置以一定速率转动,可以将电流效率由50%提高到70%。同时,树枝状电沉积产物的问题得到缓解,大量减少了脱落到液态镉层中的铀,使得电解精炼过程变的更加稳定[57]。
当电解精炼流程实施几批次后,熔盐中累积的超铀元素和稀土元素氯化物会达到很高浓度,则需要提取超铀元素。电解提取的任务是回收熔盐中累积的超铀元素(主要是钚)[58-61]。电解提取时,将固态阴极换成装在坩埚中的液态镉阴极,仍以装有乏燃料的不锈钢吊篮为阳极。通电电解后,铀和超铀元素同时沉积到镉电极中,以溶质或金属间化合物形式溶解在液态镉中,该过程不会产生纯钚,具有防止核扩散作用。由于超铀元素在液态镉中的活度大幅度减小,使得它们能够稳定存在于含有UCl3的熔盐中,不发生置换反应。但是,由于稀土元素在熔盐和液态镉中的热力学性质与次锕系元素相近,所以即使很好的控制电解条件,仍有少量的稀土元素会共沉积到液态镉中[62]。
在电解提取过程中,同样存在沉积物呈现树枝状的问题,这是由于铀合金化速率较慢,会在液态镉表面堆积,而不溶解到液态镉中生成金属间化合物,这样就会阻止其它元素电沉积到液态镉层中。为此,在设计电解装置时,需要在液态镉层中安装刮刀或搅拌器等,不断将铀从液态镉表面通过机械装置转移到液态镉中[12]。
电解精炼结束后,由于固体阴极上粘附了约20%(质量分数)的熔盐和液态阴极镉需要和锕系产物分离,ANL和INL开发了阴极产物处理装置,采用真空蒸馏法去除粘附的熔盐和分离镉。真空蒸馏的实施条件是1 200 ℃和压力小于100 Pa,容器是含有氧化锆涂层的石墨坩埚。经过真空蒸馏处理后,纯铀中氯化物盐的含量低于10 mg/kg和锕系合金铸锭中镉的含量低于35 mg/kg。回收得到的盐和镉可以返回电解精炼装置中重新使用[63-65]。
为了发展熔盐电解干法后处理技术,欧盟在21世纪初资助了高温冶金后处理研究项目(pyrometallurgical processing research programme, PYROREP),用于开展先进干法后处理技术的基础研究。在该研究计划中,欧洲各国合作开展了一系列的锕系元素和稀土元素在固态Al电极上电化学行为的基础研究[66-69],测定了析出电位等热力学参数,发现锕系元素和稀土元素在Al电极上沉积的电位差大于液态镉电极和铋电极。在此基础上,JRC-ITU提出采用固态Al电极取代液态镉电极,用于分离锕系元素和稀土元素,提高分离效率[70-71]。ITU[72]采用模拟乏燃料U60Pu20Zr10Am2Nd3.5Y0.5Ce0.5Gd0.5在固态Al电极上进行了电解精炼实验,沉积物中绝大多数是U、Pu和Am,U/Nd,Pu/Nd和Am/Nd的比例大于2 000、990和55,表明固态Al电极可以有效地提高熔盐电解法的去污因子。
近年来,我国高能物理研究所和哈尔滨工程大学提出采用AlCl3在熔盐体系中直接氯化氧化物乏燃料,然后在固态Al或W等电极上共沉积Al-An和Al-RE合金,用于分离锕系和稀土元素,也取得了较好的分离效果[73-76]。
2.4 熔盐净化和废物处理
熔盐电解干法后处理技术最终产生两种形式的核废物:金属废物和含有裂变产物的氯化物熔盐废物。金属废物主要包括包壳材料、未溶解的贵金属、残留的锕系金属和前端处理留下的金属材料。熔盐废物中的裂变产物主要包括:碱金属、碱土金属、稀土和卤化物等,其中129I和99Tc属于长寿命裂变产物,137Cs和90Sr是主要的释热核素;稀土等裂片产物属于高放射性废物[11]。金属废物较稳定,处理方法相对简单。将粘附的熔盐通过真空蒸馏分离后,向金属废物中添加适量的金属锆,融化后形成稳定的低共熔点Fe-Zr合金[77],该合金即为最终长期储存的金属核废料。熔盐废物属于高放射性废物,其中包含了大量的氯离子,其状态不稳定。因此,需要将熔盐废物转化为稳定的材料形式,然后再将其储存在与生物圈永久隔离的场所。美国ANL和INL以及韩国KAERI对熔盐电解法产生的熔盐废物的处置方法以及裂变产物的选择性分离方法进行了大量的研究。
(1) 预处理
从电解精炼装置中卸载的熔盐中含有大量的锕系元素,在进行废物处理之前,必须将这部分锕系元素回收[78]。ANL在20世纪90年代开发了从熔盐中提取锕系元素的多重还原萃取法[79]。其原理是利用锕系元素和裂变产物在液态镉相和熔盐相中的分配系数不同。当熔盐废物和含0.2%(质量分数)金属锂的液态镉进行逆流接触萃取时,稀土元素倾向于以氯化物形式存在于熔盐中,而锕系元素倾向于被金属锂还原到液态镉相中。因此,经过多次逆流萃取后,95%~99%锕系元素被还原到液态镉中,而几乎100%的稀土元素留在熔盐相中,实现了锕系元素与稀土元素的分离。为了完全回收残留的次锕系元素,需要将萃取剂换成含金属铀的液态金属镉(Cd-2%U(质量分数))。将熔盐与该萃取剂逆流接触时,残余超铀元素会被铀还原,进入液态镉相中。但是,约20%的稀土元素也被萃取到液态镉相中。通过上述多次还原萃取流程,熔盐中超铀元素的含量将小于20 mg/kg,达到了回收次锕系元素的目的。最终的熔盐废物中残留有少量铀元素,这部分铀可以被转化为废物进行最终处置;也可以采用电解的方式,将其电沉积到固态阴极上,实现回收再利用,例如ITU提出的在Al电极上彻底电解锕系元素[15]。通过整套流程,实现了锕系元素与裂变产物的分离,大大降低了熔盐中高放射性废物的含量,减小了熔盐废物的放射毒性。
(2) 制造陶瓷废物
通常水法产生的高放射性核废料采用玻璃固化的方式处理。但是由于玻璃能够容纳的氯离子量特别少,该法不适合熔盐电解法产生的高放射性废物。例如,硼硅酸玻璃可以容纳超过20%(w)的氧化钠,却只能容纳w=1%的氯离子。因此,玻璃不能够直接用于固化含大量氯离子的熔盐[11]。ANL开发了将氯化物熔盐废物制造成陶瓷材料的方法,用于固定裂变产物和微量锕系元素。该法首先将冷却的熔盐磨碎至粒径45~250 μm的颗粒,然后加入同样粒径的Zeolite-A颗粒混合均匀,加热至773 K后保温18 h,使得所有熔盐被Zeolite-A吸附,封闭进“笼形”晶体结构中。接着将吸附了熔盐的Zeolite-A冷却后与硼硅酸盐玻璃熔体颗粒按照质量比3∶1混合,加热至900 ℃以上后,沸石Zeolite-A被转化成方钠石,同时被完全封闭在陶瓷中。通过上述流程,将熔盐废物制造为耐浸出的陶瓷形式废物,适合进行永久地质储存[11,78-79]。总体来说,该流程简单易操作,是目前能够实际应用的方法,但其经济性和环境友好性还有待继续提升。首先,由于没有采取选择性分离裂变产物,导致所有LiCl-KCl共晶盐都被制造成陶瓷废物,造成高放射性陶瓷废物体积大,且LiCl-KCl熔盐无法回收再利用。此外,由于没有分离碱金属、碱土金属和稀土元素,所以无法进行分类储存[80]。近年来,为了减少高放射性熔盐废物的数量,美国ANL和INL提出采用负载了Li+和K+的Zeolite-A与熔盐废物进行离子交换,将裂变产物选择性吸附进入沸石中[81-83],从而降低熔盐中裂变产物的浓度,使得电解精炼流程可以持续进行[83]。离子交换法可以循环利用LiCl-KCl熔盐,减少了陶瓷废物的数量。
(3) 裂变产物的选择性分离
永久地质储存库的储存能力与核废料释放的衰变热速率以及总量有密切关系。由于核废料最开始阶段产生的衰变热量将最终决定高放射性废物的体积,因此提前去除核废料中的主要释热核素(137Cs和90Sr),将能够大大减小高放射性废物的体积。分离后的137Cs和90Sr可以作为非高放射性核废料,临时储存在地表处置场所[77]。稀土元素属于高放射性裂变产物,将其从熔盐中分离,可以实现熔盐回收再利用,最终达到减少高放射性废物体积的目的[84]。真空蒸馏法是常用的核废物物理分离方法。当两个组分的蒸汽压相差达到12个数量级时,可以采用真空蒸馏法实现有效地分离。然而,熔盐中以氯化物形式存在的裂变产物与熔盐蒸汽压相近,无法直接采用真空蒸馏法实现有效分离。因此,研究者希望通过将氯化物转化为其它形式无机盐或氧化物的方式,将裂变产物的蒸汽压降低,则可采用真空蒸馏法将其与熔盐分离[85]。裂变产物脱氯的方法主要包括碳酸盐和硫酸盐法(针对碱土金属)、磷酸盐沉淀法和氧化沉淀法(针对稀土元素)。
碳酸盐和硫酸盐法是由KAERI[86]提出的,用于分离LiCl熔盐体系中的碱金属和碱土金属。在LiCl熔盐体系中,首先将裂变产物铯、锶和钡转化为碳酸盐或硫酸盐,然后通过真空蒸馏法将裂变产物与熔盐体系分离。但是,CsCl不与Li2CO3反应。此外,CsCl虽然可以与Li2SO4反应,但是该反应转化率不高,最终CsCl的分离效率只达到72%。因此,碳酸盐和硫酸盐转化法不适合分离LiCl熔盐中的铯。
磷酸盐沉淀法由英国Volkovich研究小组[87]提出,可以有效地从氯化物熔盐中分离稀土裂变产物。他们根据磷酸盐稀土在氯化物熔盐中不溶的性质,将氯化物稀土转化为磷酸盐,沉淀到熔盐底部,然后再进行分离。法国[88]、英国[89]、日本[90-91]和韩国[92]也相继验证了该法的可行性。磷酸盐沉淀法也被尝试用来沉淀碱金属(Cs)和碱土金属(Sr和Ba)。结果表明,在LiCl-KCl熔盐中,CsCl、SrCl2和BaCl2无法通过添加Li3PO4和K3PO4来转化为磷酸盐。
为了分离LiCl-KCl熔盐中稀土元素裂变产物,韩国KAERI[93-95]提出先将氯化稀土转化成氧化稀土或氯氧稀土沉淀、然后再将其与熔盐分离的流程。该方法的优点是采用氧气作为氧化剂,不添加其它化学试剂,保证了不改变LiCl-KCl共晶盐的摩尔比和不产生其它杂质。向LiCl-KCl熔盐废物中缓慢通入氧气,氧气会与稀土离子反应生成稀土氧化物或氯氧化物,最终沉淀到熔盐底部,熔盐体系不会发生改变。通过控制通入氧气的速率和氧化反应的温度以及时间,稀土氯化物可以彻底反应完全。当氧化反应结束后,待稀土氧化物或氯氧化物完全沉淀到熔盐底部,快速冷却熔盐,即可得到分层的熔盐体系和沉淀产物。分离得到的稀土沉淀物中,很大一部分为氯氧化物,仍然包含了大量氯离子,不够稳定,因此,需要继续实施脱氯流程。KAERI[96]研究表明,在有氧气的条件下,将氯氧稀土加热到温度超过1 100℃时,氯氧稀土将完全转化为氧化稀土,释放出氯气。
由于CsCl性质与LiCl十分相近,脱氯法很难从熔盐中去除CsCl。此外,在650 ℃的LiCl熔盐体系中, Zeolite-A的结构已经被破坏,不再具备离子交换的能力。因此也无法采用离子交换法从LiCl熔盐体系中去除铯元素。针对上述问题,KAERI和INL[97-99]提出采用熔融结晶法(melt crystallization)从LiCl熔盐中分离碱金属和碱土金属的流程。研究表明,90%的CsCl和SrCl2杂质通过熔融结晶流程能够被富集在10%的熔盐中。因此90%的熔盐可以直接再次循环利用。剩余的10%富集了裂变产物的氯化物熔盐可以制造成陶瓷废物,进行永久地质储存。熔融结晶法不仅操作简单,而且成功地分离了熔盐体系中最难去除的Cs元素。因此,该法是需要重点研究的熔盐废物处理方法。
3 存在的问题
经过30多年的发展,基于熔盐电解法的乏燃料后处理技术得到长足发展,已经完成了全流程的实验室级别的试验,克服了大多数的困难,正迈向工程化。但是,熔盐电解技术仍然存在诸多不确定性。
第一,熔盐电解法属于干法,全流程必须在低湿低氧条件下操作,并且大多数流程都是在高温条件下完成的。换句话说,熔盐电解法面临着高温、强腐蚀、强放射性等极端条件,对整个系统的材料和操作带来极大的挑战,工程化过程中必将遇到诸多困难。第二,熔盐电解还原流程面临着严重的困难。首先,阳极铂电极消耗严重,碳材料会污染阴极产物,都不适合于工程化应用,仍需开发出合适的阳极材料。其次,电解还原流程的研究主要集中在还原氧化铀,对超铀元素氧化物熔盐还原过程的机理研究较少,还原率有待进一步研究。从有限的资料中可以看出,电解还原氧化钚的还原率和电流效率都仍然偏低,无法达到工程化要求。因此,还需大量的基础研究来改善电解还原流程。第三,熔盐电解精炼流程仍然存在难以克服的困难。电解精炼产物树枝状金属铀虽然可通过机械移除的方式得到缓解,但这不能从根本上解决短路的问题,工程化电解池操作过程中仍会不时发生短路问题。此外,机械刮刀的设计导致电解池结构极其复杂,不利于维护,也给换料等带来困难。此外,针对提取钚和次锕系元素的研究尚少,尚不能解决锕系元素与稀土元素分离的难题。即使采用固态Al电极,分离效果仍有待提升,固态Al电极能否工程化尚待考验。如果不能提高电解提取流程的去污因子,将影响后续的嬗变处理。第四,虽然目前针对熔盐废物也提出了诸多解决方案,但都只停留在模拟阶段,仍然需要持续的基础研究和工程化试验。
笔者所在实验室正在自主研发熔盐电解流程:针对枝晶金属铀沉积物问题,尝试通过添加添加剂的方法来予以解决;为提高去污因子,考察不同共沉积方法和降低操作温度对分离效率的影响;针对熔盐电解还原MOX燃料效率低等问题,正开展基础研究,包括熔盐体系的选择、电解池结构设计到计算机模拟等,希望能够给出较好的解决方案。
4 结束语
韩国已明确将熔盐电解法列为未来乏燃料后处理的首选技术,并希望到2030年能够利用该技术建立商用示范后处理工厂,使得熔盐电解干法后处理技术在韩国得到飞速发展。美国和日本都已经完成了真实乏燃料的熔盐电解法后处理实验,得到了较好的结果。日本已经完成了处理能力为40 t/a的放大实验工厂的概念设计。欧洲和印度也不断增加熔盐电解干法后处理技术的研究经费,并已经完成了实验室规模的试验。但是,我国在熔盐电解法后处理技术上仍处于起步阶段,人力、财力和物力的投入与国外还存在较大差距。
我国发展熔盐电解干法后处理技术,一定要坚持自力更生和自主创新,掌握具有自主知识产权的核心技术。笔者认为,我国需要持续大力度投入政策和资源支持熔盐电解法乏燃料后处理技术的发展,有必要将其列入国家重大科研计划中,在有条件的科研院所建立冷试验和热试验实验室,重点突破耐辐照和腐蚀的高温材料和熔盐电解流程的开发设计,大力建设高性能计算机模拟能力,完成全流程工程设计,实现全流程自动化操作,从而优选出符合我国需求的干法后处理技术路线。更重要的是,我国亟需培养一大批从事乏燃料后处理工作的专业科技人员,才能不断提高我国在该领域的核心竞争力。
致谢:感谢贾建平博士在本文成稿过程中提供的帮助和支持!
[1] 王宏渊.我国快堆闭式核燃料循环体系的现状及展望[J].能源工程,2013,5:8-12.
[2] 韦悦周,吴艳,李辉波.最新核燃料循环[M].上海:上海交通大学出版社,2016:122.
[3] 顾忠茂.我国先进核燃料循环技术发展战略的一些思考[J].核化学与放射化学,2006,28(1):1-10.
[4] 顾忠茂,柴之芳.关于我国核燃料后处理/再循环的一些思考[J].化学进展,2011,23(7):1263-1271.
[5] 韦悦周.国外核燃料后处理化学分离技术的研究进展及考察[J].化学进展,2011,23(7):1272-1288.
[6] 欧阳应根.干法后处理技术典型流程综述[M]∥中国核科技报告.2001(0):367-381.
[7] 刘丽君.国外乏燃料高温冶金后处理产生废物的处理方法[J].辐射防护,2008,28(3):184-188.
[8] 刘学刚.乏燃料干法后处理技术研究进展[J].核化学与放射化学,2009,31(增):35-44.
[9] Chang Y I. The integral fast reactor[J]. Nucl Technol, 1989, 88(2): 129-138.
[10] Laidler J J, Battles J E, Miller W E, et al. Development of pyro-processing technology[J]. Prog Nucl Energ, 1997, 31(1-2): 131-140.
[11] National Research Council. Electrometallurgical techniques for DOE spent fuel treatment: final report[M]. Washington D C: National Academy Press, 2000: 1-10.
[12] Vaden D, Li S X, Westphal B R, et al. Engineering-scale liquid cadmium cathode experiments[J]. Nucl Technol, 2008, 162(2): 124-128.
[13] Iizuka M, Koyama T, Kondo N,et al. Actinides recovery from molten salt/liquid metal system by electrochemical methods[J]. J Nucl Mater, 1997, 247(1): 183-190.
[14] Song K C, Lee H, Hur J M, et al. Status of pyroprocessing technology development in Korea[J]. Nucl Eng Technol, 2010, 42(2): 131-144.
[16] Herrmann S D, Li S X, Simpson M F, et al. Electrolytic reduction of spent nuclear oxide fuel as part of an integral process to separate and recover actinides from fission products[J]. Sep Sci Technol, 2006, 41(10): 1965-1983.
[17] Inoue T, Koch L. Development of pyroprocessing and its future direction[J]. Nucl Eng Technol, 2008, 40(40): 183-190.
[18] Lee H, Park G I, Kang K H, et al. Pyroprocessing technology development at KAERI[J]. Nucl Eng Technol, 2011, 43(4): 317-328.
[19] Vavilov S, Kobayashi T, Myochin M. Principle and test experience of the RIAR’s oxide pyro-process[J]. J Nucl Sci Technol, 2004, 41(10): 1018-1025.
[20] Bateman K J, Westphal B R, Stawicki M A. Experimental equipment design and testing of the DEOX process: Paper No. ICONE12-49451[C]∥Amercan Society of Mechanical Engineers. 12th International Conference on Nuclear Engineering, April 25-29, 2004, Arlington, Virginia, USA. 2004: 351-356.
[21] Jung I H, Shin J M, Park J J, et al. Design report:an off gas trapping system for a voloxidizer in INL of US, KAERI/TR-3245/2006[R]. Korea: KAERI, 2006.
[22] Westphal B R, Bateman K J, Lind R P, et al. Fission product removal from spent oxide fuel by head-end processing: Proceedings of Global 2005, AESJ, 2005, Tsukuba, Japan[C]. USA: Idaho National Laboratory, 2005.
[23] Yoo J H, Seo C S, Kim E H, et al. A conceptual study of pyroprocessing for recovering actinides from spent oxide fuels[J]. Nucl Eng Technol, 2007, 40(7): 581-592.
[24] Westphal B R, Bateman K J, Morgan C D, et al. Effect of process variables during the head-end treatment of spent oxide fuel[J]. Nucl Technol, 2008, 162(2): 153-157.
[25] Westphal B R, Park J J, Shin J M, et al. Selective trapping of volatile fission products with an off-gas treatment system[J]. Sep Sci Technol, 2008, 43(9-10): 2695-2708.
[26] Wahlquist D L, Brian K J B, Westphal R. Second generation experimental equipment design to support voloxidation testing at INL: 16th International Conference on Nuclear Engineering (ICONE16), May 11-15, 2008, Orlando, Florida, USA[C]. Orlando, 2008.
[27] Chen G Z, Fray D J, Farthing T W. Direct electrochemical reduction of titanium dioxide to titanium in molten calcium chloride[J]. Nature, 2000, 407(6802): 361-364.
[28] Herrmann S D, Li S X, Simpson M F, et al. Electrolytic reduction of spent oxide fuel bench-scale test preparations: Proceedings of ANS Fifth Topical Meeting on DOE Spent Nuclear Fuel and Fissile Materials Management, Charleston, USA, September 17-20, 2002[C]. Charleston, 2002.
[29] Gourishankar K, Redey L, Williamson M. Electrolytic reduction of metal oxides in molten salts[M]. Schneider W A. Light Metals 2002. Warrendale, Pennsylvania: The Minerals, Metals, and Materials Society, 2002: 1075-1082.
[30] Hur J M, Seo C S, Hong S S, et al. Metallization of U3O8viacatalytic electrochemical reduction with Li2O in LiCl molten salt[J]. React Kinet Catal Lett, 2003, 80(2): 217-222.
[31] Sakamura Y, Omori T. Electrolytic reduction and electrorefining of uranium to develop pyrochemical reprocessing of oxide fuels[J]. Nucl Technol, 2008, 171(3): 266-275.
[32] Seo C S, Park S B, Park B H, et al. Electrochemical study on the reduction mechanism of uranium oxide in a LiCl-Li2O molten salt[J]. J Nucl Sci Technol, 2006, 43(5): 587-595.
[33] Jeong S M, Shin H S, Hong S S, et al. Electrochemical reduction behavior of U3O8powder in a LiCl molten salt[J]. Electrochim Acta, 2010, 55(5): 1749-1755.
[34] Kurata M, Inoue T, Serp J, et al. Electro-chemical reduction of MOX in LiCl[J]. J Nucl Mater, 2004, 328(2-3): 97-102.
[35] Iizuka M, Sakamura Y, Inoue T. Electrochemical reduction of (U-40Pu-5Np)O2in molten LiCl electrolyte[J]. J Nucl Mater, 2006, 359(1-2): 102-113.
[36] Iizuka M, Inoue T, Ougier M, et al. Electrochemical reduction of (U, Pu)O2in molten LiCl and CaCl2electrolytes[J]. J Nucl Sci Technol, 2007, 44(5): 801-813.
[37] Vishnu D S M, Sanil N, Murugesan N, et al. Determination of the extent of reduction of dense UO2cathodes from direct electrochemical reduction studies in molten chloride medium[J]. J Nucl Mater, 2012, 427(1-3): 200-208.
[38] Vishnu D S M, Sanil N, Panneerselvam G, et al. Mechanism of direct electrochemical reduction of solid UO2to uranium metal in CaCl2-48mol% NaCl melt[J]. J Electrochem Soc, 2013, 160(9): D394-D402.
[39] Choi E Y, Hur J M, Choi I K, et al. Electrochemical reduction of porous 17 kg uranium oxide pellets by selection of an optimal cathode/anode surface area ratio[J]. J Nucl Mater, 2011, 418(1-3): 87-92.
[40] Choi E Y, Im H S, Hur J M. Effect of the anode-to-cathode distance on the electrochemical reduction in a LiCl-Li2O molten salt[J]. J Korean Electrochem Soc, 2013, 16(3): 138-144.
[41] Choi E Y, Kim J K, Im H S, et al. Effect of the UO2form on the electrochemical reduction rate in a LiCl-Li2O molten salt[J]. J Nucl Mater, 2013, 437(1-3): 178-187.
[42] Park W, Kim J K, Hur J M, et al. Application of a boron doped diamond (BDD) electrode as an anode for the electrolytic reduction of UO2in Li2O-LiCl-KCl molten salt[J]. J Nucl Mater, 2013, 432(1-3): 175-181.
[43] Choi E Y, Won C Y, Cha J S, et al. Electrochemical reduction of UO2in LiCl-Li2O molten salt using porous and nonporous anode shrouds[J]. J Nucl Mater, 2014, 444(1-3): 261-269.
[44] Herrmann S D, Li S X. Separation and recovery of uranium metal from spent light water reactor fuel via electrolytic reduction and electrorefining[J]. Nucl Technol, 2010, 171(3): 247-265.
[45] Herrmann S D, Li S X, Westphal B R. Separation and recovery of uranium and group actinide products from irradiated fast reactor MOX fuel via electrolytic reduction and electrorefining[J]. Sep Sci Technol, 2012, 47(14-15): 2044-2059.
[46] Simpson M F, Herrmann S D. Modeling the pyrochemical reduction of spent UO2fuel in a pilot-scale reactor[J]. Nucl Technol, 2008, 162(2): 179-183.
[47] Phongikaroon S, Herrmann S D, Simpson M F. Diffusion model for electrolytic reduction of uranium oxides in a molten LiCl-Li2O salt[J]. Nucl Technol, 2011, 174(1): 85-93.
[48] Jeong S W, Park S B, Hong S S, et al. Electrolytic production of metallic uranium from U3O8in a 20 kg-batch scale reactor[J]. J Radioanal Nucl Chem, 2006, 268(2): 349-356.
[49] Choi E Y, Lee J W, Park J J, et al. Electrochemical reduction behavior of a highly porous SIMFUEL particle in a LiCl molten salt[J]. Chem Eng J, 2012, 207-208: 514-520.
[50] Sakamura Y, Akagi M. Pyrochemical reprocessing tests to collect uranium metal from simulated spent oxide fuel[J]. Nucl Technol, 2012, 179(2): 220-233.
[51] Hur J M, Choi I K, Cho S H, et al. Preparation and melting of uranium from U3O8[J]. J Alloys Compd, 2008, 452(1): 23-26.
[52] Simpson M F. Developments of spent nuclear fuel pyroprocessing technology at idaho national laboratory, INL/EXT-12-25124[R]. Idaho: Idaho National Laboratory, 2012.
[53] Battle J E, Laidler J J, McPheeters C C, et al. Pyrometallurgical processes for recovery of actinide elements, ANL/CMT/CP—81050, CONF-940215-4[R]. USA: ANL, 1994.
[54] Benedict R, Goff M, Teske G, et al. Progress in electrometallurgical treatment of spent nuclear fuel[J]. Nucl Sci Technol, 2002, 39(Sup 3): 749-752.
[55] Li S X, Vaden D, Mariani R D. Experimental observations on the role of the cadmium pool in mark-Ⅳ ER: Proceedings of Topical Meeting of DOE Spent Nuclear Fuel and Fissile Material Management, June 4-8, 2000, San Diego, CA[C]. San Diego, 2000.
[56] Ahluwalia R K, Hua T Q, Vaden D. Uranium transport in a high-throughput electrorefiner for EBR-Ⅱ blanket fuel[J]. Nucl Technol, 2004, 145(1): 67-81.
[57] Li S X, Johnson T A, Westphal B R, et al. Electrorefining experience for pyrochemical processing of spent EBR-Ⅱ Driver Fuel: Proceedings of GLOBAL 2005, Paper No. 487, Tsukuba, Japan, 2005[C]. Tsukuba, 2005.
[58] Koyama T, Iizuka M, Shoji Y, et al. An experimental study of molten salt electrorefining of uranium using solid iron cathode and liquid cadmium cathode for development of pyrometallurgical reprocessing[J]. J Nucl Sci Technol, 1997, 34(4): 384-393.
[59] Kinoshita K, Inoue T, Fusselman S P, et al. Electrodeposition of uranium and transuranic elements onto solid cathode in LiCl-KCl/Cd system for pyrometallurgical partitioning[J]. J Nucl Sci Technol, 2003, 40(7): 524-530.
[60] Li S X, Herrmann S D, Benedict R W, et al. Actinide recovery experiments with bench-scale liquid cadmium cathode in real fission product-laden molten salt[J]. Nucl Technol, 2009, 165(2): 190-199.
[61] Li S X, Herrmann S D, Simpson M F. Electrochemical analysis of actinides and rare earth constituents in liquid cadmium cathode product from spent fuel electrorefining[J]. Nucl Technol, 2010, 71(3): 292-299.
[62] Li S X, Herrmann S D, Simpson M F. Experimental investigations into U/TRU recovery using a liquid cadmium cathode and salt containing high rare earth concentrations: Proceedings of GLOBAL 2009, Paris, France, September 6-11, 2009[C]. Paris, 2009.
[63] Westphal B R, Vaden D, Hua T Q, et al. Recent developments at the cathode processor for spent fuel treatment[C]. Proceedings of American Nuclear Society Fifth Topical Meeting, DOE Spent Nuclear Fuel and Fissile Materials Management, Charleston, South Carolina, USA, 2002.
[64] Westphal B R, Price J C, Vaden D, et al. Engineering-scale distillation of cadmium for actinide recovery[J]. J Alloys Compd, 2007, 444(445): 561-564.
[65] Westphal B R, Marsden K C, Price J C, et al. On the development of a distillation process for the electrometallurgical treatment of irradiated spent nuclear fuel[J]. Nucl Eng Technol, 2008, 40(3): 163-174.
[66] Castrillejo Y, Bermejo R, Martínez A M, et al. Application of electrochemical techniques in pyrochemical processes - electrochemical behavior of rare earths at W, Cd, Bi and Al electrodes[J]. J Nucl Mater, 2007, 360(1): 32-42.
[67] Cassayre L, Caravaca C, Jardin R, et al. On the formation of U-Al alloys in the molten LiCl-KCl eutectic[J]. J Nucl Mater, 2008, 378(1): 79-85.
[69] Mendes E, Malmbeck R, Nourry C, et al. On the electrochemical formation of Pu-Al alloys in molten LiCl-KCl[J]. J Nucl Mater, 2012, 420(1-3): 424-429.
[70] Serp J, Allibert M, Terrier A L, et al. Electroseparation of actinides from lanthanides on solid aluminum electrode in LiCl-KCl eutectic melts[J]. J Electrochem Soc, 2005, 152(3): C167-C172.
[71] Conocar O, Douyere N, Glatz J-P, et al. Promising pyrochemical actinide/lanthanide separation processes using aluminum[J]. Nucl Sci Eng, 2006, 153(3): 253-261.
[72] Serp J, Malmbeck R, Scheppler C, et al. Electroseparation of actinide using Al cathodes in LiCl-KCl: Proceedings of Atalante[C]. Nimes, France, June 21-24, 2004: 365-371.
[73] Liu Y L, Yan Y D, Han W, et al. Extraction of thorium from LiCl-KCl molten salts by forming Al-Th alloys: a new pyrochemical method for the reprocessing of thorium-based spent fuels[J]. RSC Adv, 2013, 3(45): 23539-23547.
[74] Liu Y L, Yan Y D, Han W, et al. Electrochemical separation of Th from ThO2and Eu2O3assisted by AlCl3in molten LiCl-KCl[J]. Electrochim Acta, 2013, 114: 180-188.
[75] Liu Y L, Ye G A, Yuan L Y, et al. Electroseparation of thorium from ThO2and La2O3by forming Th-Al alloys in LiCl-KCl eutectic[J]. Electrochim Acta, 2015, 158: 277-286.
[76] Tang H, Yan Y D, Zhang M L, et al. AlCl3-aided extraction of praseodymium from Pr6O11in LiCl-KCl eutectic melts[J]. Electrochim Acta, 2013, 88: 457-462.
[77] Keiser Jr D D, Abraham D P, Sinkler W, et al. Actinide distribution in a stainless steel-15 wt% zirconium high-level nuclear waste form[J]. J Nucl Mater, 2000, 279(2-3): 234-244.
[78] Priebe S, Bateman K. The ceramic waste form process at the Idaho National Laboratory[J]. Nucl Technol, 2008, 162(2): 199-207.
[79] Ackerman J P, Johnson T R, Chow L S H, et al. Treatment of wastes in the IFR fuel cycle[J]. Prog Nucl Energy, 1997, 31(1-2): 141-154.
[80] Simpson M F, Yoo T S, Benedict R W, et al. Strategic minimization of high level waste from pyroprocessing of spent nuclear fuel: Proceedings of GLOBAL 2007, 1394-1397, Boise, USA, September 9-13, 2007[C]. USA: Idaho National Laboratory, 2007.
[81] Phongikaroon S, Simpson M F. Equilibrium model for ion exchange between multivalent cations and zeolite-a in a molten salt[J]. AIChE J, 2006, 52(5): 1736-1743.
[82] Yoo T S, Frank S M, Simpson M F, et al. Salt-zeolite ion-exchange equilibrium studies for a complete set of fission products in molten LiCl-KCl[J]. Nucl Technol, 2010, 171(3): 306-315.
[83] Shaltry M, Phongikaroon S, Simpson M F. Ion exchange kinetics of fission products between molten salt and zeolite-A[J]. Micropor Mesopor Mater, 2012, 152: 185-189.
[84] Eun H C, Cho Y Z, Son S M, et al. Recycling of LiCl-KCl eutectic based salt wastes containing radioactive rare earth oxychlorides or oxides[J]. J Nucl Mater, 2012, 420(1-3): 548-553.
[85] Cho Y J, Yang H C, Eun H C, et al. Oxidation of lanthanum chloride in a LiCl-KCl eutectic molten salt using the oxygen gas sparging method[J]. J Ind Eng Chem, 2005, 11(5): 707-711.
[86] Eun H C, Cho Y Z, Park H S, et al. Study on a separation method of radionuclides (Ba, Sr) from LiCl salt wastes generated from the electroreduction process of spent nuclear fuel[J]. J Radioanal Nucl Chem, 2012, 292(2): 531-535.
[87] Volkovich V A, Griffiths T R, Thied R C. Treatment of molten salt wastes by phosphate precipitation: removal of fission product elements after pyrochemical reprocessing of spent nuclear fuels in chloride melts[J]. J Nucl Mater, 2003, 323(1): 49-56.
[88] Hudry D, Bardez I, Rakhmatullin A, et al. Synthesis of rare earth phosphates in molten LiCl-KCl eutectic: application to preliminary treatment of chlorinated waste streams containing fission products[J]. J Nucl Mater, 2008, 381(3): 284-289.
[89] Harrison M T, Simms H E, Jackson A, et al. Salt waste treatment from a LiCl-KCl based pyrochemical spent fuel treatment process[J]. Radiochim Acta, 2008, 96(4-5): 295-301.
[90] Amamoto I, Kofuji H, Myochin M, et al. Phosphates behaviours in conversion of FP chlorides[J]. J Nucl Mater, 2009, 389(1): 142-148.
[91] Amamoto I, Kofuji H, Myochin M, et al. Precipitation behaviors of fission products by phosphate conversion in LiCl-KCl medium[J]. Nucl Technol, 2010, 171(3): 316-324.
[92] Cho Y Z, Lee T K, Eun H C, et al. Purification of used eutectic (LiCl-KCl) salt electrolyte from pyroprocessing[J]. J Nucl Mater, 2013, 437(1-3): 47-54.
[93] Cho Y J, Yang H C, Eun H C, et al. Characteristics of oxidation reaction of rare-earth chlorides for precipitation in LiCl-KCl molten salt by oxygen sparging[J]. J Nucl Sci Technol, 2006, 43(10): 1280-1286.
[94] Eun H C, Yang H C, Cho Y J, et al. Separation of pure LiCl-KCl eutectic salt from a mixture of LiCl-KCl eutectic salt and rare-earth precipitates by vacuum distillation[J]. J Nucl Sci Technol, 2007, 44(10): 1295-1300.
[95] Cho Y Z, Park G H, Yang H C, et al. Minimization of eutectic salt waste from pyroprocessing by oxidative precipitation of lanthanides[J]. J Nucl Sci Technol, 2009, 46(10): 1004-1011.
[96] Eun H C, Cho Y Z, Park H S, et al. Study on a recovery of rare earth oxides from a LiCl-KCl-RECl3system[J]. J Nucl Mater, 2011, 408(1): 110-115.
[97] Lee H S, Oh G H, Lee Y S, et al. Concentrations of CsCl and SrCl2from a simulated LiCl salt waste generated by pyroprocessing by using czochralski method[J]. J Nucl Sci Technol, 2009, 46(4): 392-397.
[98] Cho Y Z, Park G H, Lee H S, et al. Concentration of cesium and strontium elements involved in a LiCl waste salt by a melt crystallization process[J]. Nucl Technol, 2010, 171(3): 325-334.
[99] Williams A N, Phongikaroon S, Simpson M F. Separation of CsCl from a ternary CsCl-LiCl-KCl salt via a melt crystallization technique for pyroprocessing waste minimization[J]. Chem Eng Sci, 2013, 89(4): 258-263.
DevelopmentofPyroprocessingofSpentNuclearFuelbyMoltenSaltsElectrolysis
TANG Hao, REN Yi-ming, SHAO Lang, ZHONG Yi, GAO Rui
Institute of Materials, China Academy of Engineering Physics, Jiangyou 621907, China
Pyroprocessing, based on molten salts electrolysis, is the most promising technology for dry reprocessing of different types of spent fuels, such as oxide and metallic fuels. Pyroprocessing technology involves four main processes, which are head-end treatment, electrochemical reduction, electrorefining and electrowinning, and waste treatment. This paper provides a state of the art review of the basic processes and unresolved key issues of pyroprocessing technology.
spent fuel; dry reprocessing; nuclear waste; molten salts electrolysis; actinides
2016-05-17;
2016-10-08
国家自然科学基金青年科学基金资助项目(215011546;21507118)
唐 浩(1988—),男,江苏盱眙人,博士,助理研究员,主要从事核燃料干法后处理技术研究,E-mail: haotang@caep.cn
TL99
A
0253-9950(2017)06-0385-12
10.7538/hhx.2017.39.06.0385
猜你喜欢
氯碱工业(2021年5期)2021-09-10
山西化工(2020年6期)2021-01-10
今日农业(2020年20期)2020-12-15
中学生数理化(高中版.高考数学)(2020年2期)2020-04-21
中国化肥信息(2020年8期)2020-03-19
陶瓷学报(2019年6期)2019-10-27
能源(2018年10期)2018-12-08
资源节约与环保(2018年1期)2018-02-08
中国新技术新产品(2017年4期)2017-03-04
能源(2016年10期)2016-02-28
