
基于GPU的生物大分子计算平台的构建与优化
2017-12-14 02:59:38宁璐璐
食品与生物技术学报 2017年10期
俞 伟,宁璐璐, 许 菲*
(1.江南大学,信息化建设与管理中心,江苏 无锡214122;2.江南大学,生物工程学院,江苏 无锡 214122)
基于GPU的生物大分子计算平台的构建与优化
俞 伟1,宁璐璐2, 许 菲*2
(1.江南大学,信息化建设与管理中心,江苏 无锡214122;2.江南大学,生物工程学院,江苏 无锡 214122)
通过生物大分子计算平台对分子动力学的模拟运算,计算值可以描述分子的运动轨迹,从而揭示原子间的内在关系,但这一过程较为复杂且昂贵。作者研究了较低投入下大分子计算平台的构建与优化,在并行GPU条件和CUDA体系下使用AMBER软件包完成分子动力学的模拟运算。通过对两个大分子体系的运算,实验结果表明计算平台达到了60倍的计算加速,实现了较高的加速性能,完成了高性价比的高性能计算平台的建设。
生物大分子,分子动力学,计算加速,图形处理单元
随着计算机计算能力的快速提升,生物大分子的动力学模拟计算受到了科研人员的重视。分子动力学(Molecular Dynamics,MD)通过软件模拟计算出生物大分子在原子水平上的相互作用力,从而展示生物大分子运动的微观过程,得到生物大分子结构变化、系统静态和动态的动力学性质和热力学性质等详细信息。
2007年,马国正等[1]实现了双CPU下生物大分子分子动力学的模拟。2008年,Yasuda[2]最早实现了使用GPU来提高计算电子排斥积分的速度并加以改进。文献[3-5]均提及GPU对于分子动力学的模拟计算有显著的加速效果。2007年,CUDA被用于对分子动力学的计算进行改进,在GPU上计算简化的范德华势。上述研究表明多种分子计算程序包伴随着GPU的加入,计算速度得到显著的提升[6]。
综上所述,GPU的引入为计算化学带来了快速发展,基于GPU的开源软件也有了长足的进步。但是习惯于传统CPU计算化学的国内用户对于GPU还是不甚了解,甚至认为GPU是高贵神秘的技术。因此GPU在计算化学中的使用需要宣传、推广和普及。研究人员提出了使用PDB代码、Web程序等生物信息学方法描述源蛋白大分子中连接肽的位置、长度、溶剂可及性和二级结构等信息[6]。部分学科需要通过生物大分子计算平台的计算值来描述分子的运动轨迹,从而揭示原子间的内在关系,但是巨大的计算量延缓了科研的进展。作为一名计算机工作人员,协助这类学科构建大分子计算平台是作者的一项研究课题。通过实践,作者证明了可以通过较低的投入完成高性能大分子计算平台的构建。
如何构建和优化基于GPU的大分子计算平台是作者研究的重点和核心,通过对分子动力学的模拟计算,证明并行效果良好,GPU环境下计算速度较CPU环境下有近60倍的提高,为构建分子动力学计算平台提供了性价比高的解决方案。
1 相关技术
1.1AMBER
目前分子动力学计算模拟软件中,常用的包括AMBER、CHARMM和GROMOS等。其中AMBER分子力场由加利福尼亚大学P.A.Kollman课题组最初为计算蛋白质和核酸体系而开发,计算参数数据均来源于实验值。随着Kollman课题组和其他课题组对AMBER内容的不断丰富,逐渐形成了一个具备用于生物大分子、有机小分子和高分子模拟计算功能的力场体系。AMBER包含了分子动力学模拟计算的源代码和演示程序,是一个在生物大分子模拟计算领域被广泛应用的分子力场软件。AMBER的优势在于对生物大分子的计算,对小分子体系的计算还需借助其他模拟计算体系。
AMBER力场的势能函数形式简单,所需参数少,计算量比较小,但也在一定程度上限制了这个力场的扩展性。AMBER力场用傅立叶级数的形式描述二面角扭转能,用谐振子模型计算键长伸缩能和键角弯转能,用Lennard-Jones势模拟范德华力,其势能表达式(1)为:
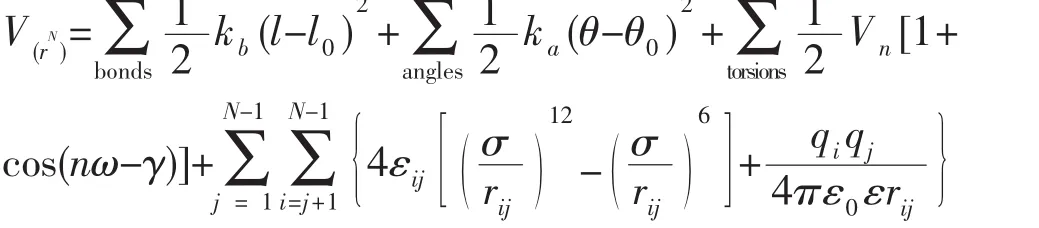
表达式(1)rij为两个原子之间的距离,σ为碰撞常数(collision parameter),εij为势阱深度(well-depth),qi和qj分别为两个原子的电荷,ε0为常数,ε为介电常数。
作者使用的计算软件AMBER 14由AMBER 12升级而来。通过升级,实现了对GPU的完全支持,将注意力集中于扩展封装性能、提升单GPU运行性能 30%、支持 CUDA(5、6和 7)、支持多 GPU 有效运行、进一步提升对分子动力学的加速等方面。
1.2CUDA在GPU计算中的使用
GPU 不仅用于图形的渲染,由于其具备万亿次浮点运算的性能导致从金融到医药产生了爆发式的应用。使用高层语言,可以在CPU上通过使用GPU加速应用的运行,既优化了CPU的单线程性能,又加速了GPU的并行处理。
CUDA是由NVIDIA公司开发的编程模型和并行计算平台,通过GPU的使用实现计算能力的大幅提高。CUDA被广泛应用于各类计算应用和研究论文,目前至少有3亿个支持CUDA的GPU被用于笔记本、工作站、计算机集群和超级计算机。通过CUDA实现了使用C、C++和Fortran等高级语言直接操作GPU,而不再需要汇编语言。
1.3 MPI的使用
在多处理器前提下,计算机指令的执行分为串行和并行两种方式。并行执行指令的方式(MPI)在完善的资源调度和通信机制下可以取得更高的性能,通过MPI可以将计算任务分摊给多个处理单元实现处理单元的负载均衡,最终实现各处理单元性能的最大发挥。MPI不是一种语言,是一种并行执行的思想,是一种规范和模型。课题使用的服务器配置了2块GPU卡,因此需要通过MPI的引入来实现2块GPU卡的并行工作,在较短的时间内完成更大的计算量。课题通过OpenMPI开源工具来实现MPI。
2 大分子计算平台的构建
针对课题计算的需求,构建了如表1的硬件平台。
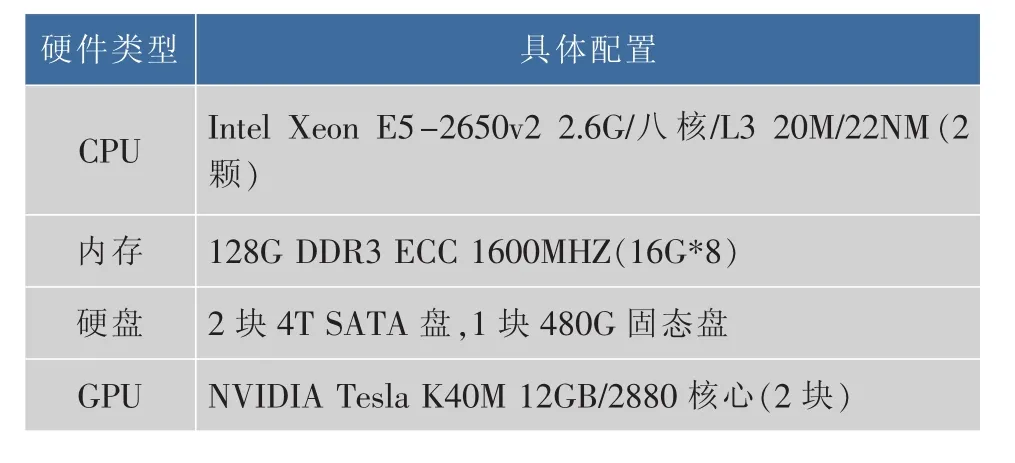
表1 计算平台硬件配置Table 1 Hardware configuration of computing platform
课题构建的生物大分子计算平台使用了NVIDIA的高性能GPU加速器TeslaK40,该加速器峰值双精度浮点性能为1.43 Tflops,峰值单精度浮点性能为4.29 Tflops,存储器带宽 (ECC关闭)为288 GB/秒,存储器容量(GDDR5)达到 12 GB,在桌面获得集群的性能。在表1的硬件条件下在CentOS 6操作系统中首先要完成基础运行环境的构建,主要包括如下两个步骤:
1.安装Intel CPU环境下的C++环境
cd/home/tony
tar zxvf l_ccompxe_2013_sp1.0.080.tgz
cd./intel/l_ccompxe_2013_sp1.0.080
su
setenforce 0
getenforce
yum install libstdc++.so.5
exit
./install.sh
cd
vim.bashrc
export MKL_HOME=/home/tony/intel/mkl
source/ home/tony/intel/composer_xe_2013_sp1.0.080/bin/compilervars.sh intel64
source .bashrc
su
yum install gcc flex tcsh zlib-devel bzip2-devel libXt-devel libXext-devel libXdmcp-devel
2. 安装分子计算平台AMBER 14 和AMBERtools 14
cd /home/tony
tar xvfj AmberTools15.tar.bz2
tar xvfj Amber14.tar.bz2
vim .bashrc
export AMBERHOME=/home/tony/amber14
source .bashrc
cd $AMBERHOME
./configure intel
make install
make test
上述环境搭建后已可以进行分子动力学的计算,但为了实现高效和并行,需对平台做如下优化,主要包括如下两个步骤:
1.编译安装openmpi-1.6.tar.bz2
tar xvfj openmpi-1.6.tar.bz2
cd /home/tony/openmpi-1.6
./configure --prefix =/home/tony/openmpi -intel
FC=ifort CC=icc CXX=icpc F77=ifort
make
make install
vim .bashrc
export MPI_HOME=/home/tony/openmpi-intel
export PATH = $MPI_HOME/bin:
$AMBERHOME/bin:$PATH
export LD_LIBRARY_PATH = $MPI_HOME/lib:
$AMBERHOME/lib:$ LD_LIBRARY_PATH
export MKL_HOME=/home/tony/intel/mkl
2.安装并行
cd $AMBERHOME
./configure-mpi intel
make install
3 实验与仿真
3.1 测试体系说明
平台对两个体系进行了测试,体系参数如表2所示。

表2 测试体系Table 2 Test system
两个体系的模拟形状如图1所示。
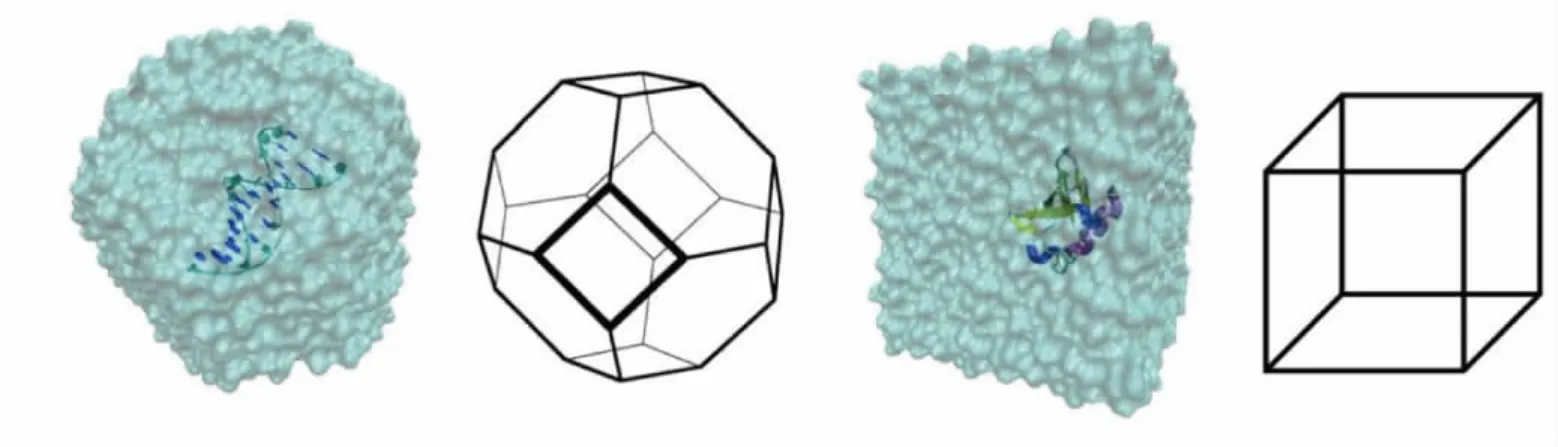
图1 DNA与蛋白质测试体系的模拟形状Fig.1 Shape of DNA and protein about simulation test system
测试体系的模拟运算参数如表3和表4所示。

表3 模拟参数Table 3 Simulation parameters

表4 力场参数Table 4 Force field parameters
3.2 实验结果
为提高计算精度,计算数据使用了双精度数据。在计算平台上分别使用单核CPU、八核CPU、单GPU和双GPU对上述DNA和蛋白质体系进行了测试。DNA测试结果如图2所示,蛋白质体系测试结果如图3所示。
在单CPU、8CPU、1GPU和2GPU条件下对于两个生物大分子体系的运算加速比如表5所示。

图2 DNA体系测试结果Fig.2 Result of DNA system
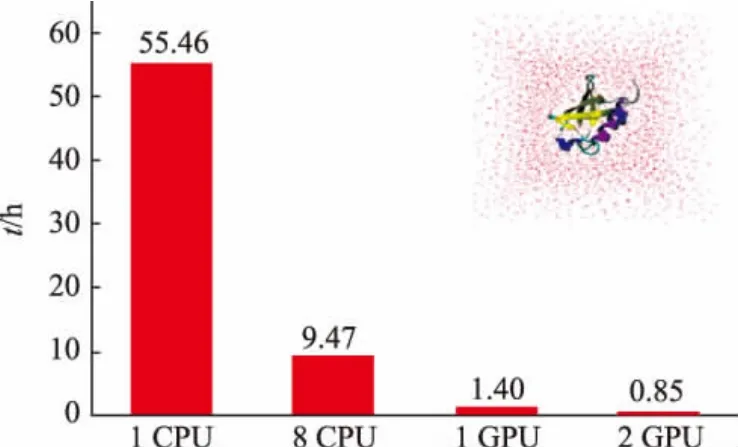
图3 蛋白质体系测试结果Fig.3 Result of protein system
表5 不同运算单元的加速比
Table 5 Speedup different process unit

2GPU57.7465.25
4 结语
实现了低投入高性价比的生物大分子计算平台的构建与优化。证明了GPU对于加速分子动力学的模拟运算起着至关重要的作用。
后续课题将从事如下两方面的工作:目前2GPU较1GPU的加速比分别是1.60和1.65,需要优化GPU环境下的计算平台,实现2GPU较1GPU的加速比接近或达到2;构建低成本的计算集群,通过多机的并行运行实现更高的加速比,为更高级别的原子数量的模拟计算提供计算平台。
[1]MA Guozheng,NAN Junmin.Molecular dynamic simulations of biomacromolecule on dual Process-based system using MPICH technology[J].Computers and Applied Chemistry,2007,24(8):852-855.(in Chinese)
[2]YASUDA K.Two-electron integral evalution on the Graphics[J].J Comput Chem,2008,29:334-342.
[3]FEI Hui,ZHANG Yunquan,WANG Ke,et al.Parallel algorithm and implementation for molecular dynamics simulation based on GPU[J].Computer Science,2011,38(9):1056-1058.(in Chinese)
[4]LIN Jianghong,LIN Jinxian,LU Tun.Accelerated molecular dynamics simulation using multi-core CPU and GPU[J].Journal of Computer Applications,2011,31(3):367-381.(in Chinese)
[5]LI Jiangyu,ZHAO Dongsheng,WANG Yumin.GPU computing and its application in biomedical research[J].Mil Med Sci,2011,35(8):1024-1028.(in Chinese)
[6]LI Jianfang,WANG Chunjuan,WU Minchen.Design of linker peptides and its application in fusion protein[J].Journal of Food Science and Biotechnology,2015,34(11).(in Chinese)
Construction and Optimization of GPU-Based Computing Platform for Biological Macromolecules
YU Wei1,NING Lulu2,XU Fei*2
(1.NIC,Jiangnan University,Wuxi,214122,China;2.SchoolofBiotechnology,Jiangnan University,Wuxi 214122,China)
Biological macromolecules by molecular dynamics computing platform for analog operation,calculated value can describe the trajectory of molecules,which reveals the intrinsic relationship between atoms,but the process is more complicated and expensive.In this paper,the construction and optimization to achieve lower investment macromolecules computing platforms,using a software package AMBER molecular dynamics simulation runs in parallel GPU and CUDA system conditions.Through the operation of two large molecules,experimental results show that the computing platform reached 60 times the computing speed up to achieve a high acceleration performance,completed the construction of cost-effective,high-performance computing platform.
biological macromolecules,molecular dynamics (MD),accelerated computing,graphic processing unit(GPU)
TP 302.7
A
1673—1689(2017)10—1101—05
2015-10-08
江苏省自然科学基金项目(BK20151126)。
俞 伟(1975—),男,江苏无锡人,工学硕士,讲师,主要从事计算机工程研究。E-mail:yuwei@jiangnan.edu.cn
*通信作者:许 菲(1978—),女,黑龙江哈尔滨人,工学博士,教授,博士研究生导师,主要从事生物化学、生物大分子研究。
E-mail:feixu@jiangnan.edu.cn
俞伟,宁璐璐,许菲.基于GPU的生物大分子计算平台的构建与优化[J].食品与生物技术学报,2017,36(10):1101-1105.
猜你喜欢
航空材料学报(2023年6期)2023-12-18 05:23:50
小学生学习指导(小军迷联盟)(2023年3期)2023-03-27 09:22:44
中国音乐学(2022年1期)2022-05-05 06:48:46
能源工程(2022年1期)2022-03-29 01:06:26
物理学报(2018年10期)2018-06-14 06:31:32
浙江大学学报(理学版)(2016年6期)2016-12-15 03:15:03
中国塑料(2016年9期)2016-06-13 03:18:54
分析测试学报(2015年3期)2016-01-13 06:18:12
中国药业(2014年17期)2014-05-26 09:07:45
火炸药学报(2014年3期)2014-03-20 13:17:43
