
一种新型液晶化合物的合成和表征
2017-12-14 01:20
分析仪器 2017年6期
(神华包头煤化工有限责任公司,包头 014010)
一种新型液晶化合物的合成和表征
王玉辉刘尚清赵灵春王涛
(神华包头煤化工有限责任公司,包头 014010)
通过醚化、还原、重氮化、偶合反应及酯化等反应,合成了新型液晶化合物4-((4-己烷氧苯基)偶氮基)苯基异烟酸酯,并将此化合物作为质子受体,以烷氧基苯甲酸为质子供体,最后合成了一种氢键型液晶复合物。用IR、1H NMR对中间体及其产物进行了结构表征,并用带热台的偏光显微镜对各产物的液晶行为进行了研究。通过偏光显微镜说明氢键复合物是典型的热致液晶且呈现明显的近晶相和向列相两种相态,并且超分子液晶复合物较质子供体的液晶相范围宽,说明分子间氢键起到了稳定液晶相态的作用。
偶氮型 近晶相 向列相
偶氮苯类液晶材料因具有光致异构,其在光记录存储、光电子器件以及全息照相等方面有广阔的应用价值。目前随着人们对光信息处理、光讯通信等领域的深入研究,非线性光学材料的地位也越来越重要[1,2]。由于偶氮苯类液晶非线性光学材料在加工和成本方面比传统的非线性光学材料有较大的优势,因此有着巨大的潜力。迄今为止,偶氮液晶化合物已经获得了重要的发展,尤其是在偶氮液晶聚合物方面的研究[3-5],取得了很大的发展。
氢键是生物体内广泛存在的一种作用力,具有饱和性和方向性。90年代,Kato 和Fréchet 等[6-8]合成了一系列氢键组装型液晶引起了广泛的关注[9-11]。氢键具有较好的组合柔性,其选择空间大,组合方式多,虽然较共价键键能弱,将会给复合物的稳定性带来不利的影响,但是它的适当弱化,增大了分子的柔性,反而使得氢键液晶复合物具有独特的动力学性质,尤其是对温度的变化,具有可逆响应性能,使其在感应材料制备方面提供了更加广阔的应用空间。
本实验以烷氧基苯甲酸为质子供体,偶氮类吡啶衍生物为质子受体,合成了一种新型氢键液晶,对所得复合物的结构和液晶性能进行了研究。合成路线如图1。

图1 新型氢键液晶合成路线
1 实验部分
1.1 实验试剂及仪器
溴代正己烷,化学纯;异烟酸,化学纯;对硝基苯酚,化学纯;N,N’-二环己基碳二亚胺(DCC,化学纯;对二甲氨基吡啶(DMAP),化学纯;有机溶剂使用前经过常规纯化,纯化方法为根据试剂沸点进行蒸馏提纯。
带热台Eclipse80i 偏光显微镜 (日本岛津公司);NIKON数码相机 (日本);(VERTEX -70)IR红外光谱仪 (美国PE公司,KBr压片);X-4显微熔点测定仪 (上海精密科学仪器有限公司);Avanci-D2X-200型核磁共振仪 (美国瓦里安公司,TMS为内标)。
1.2 中间体及其目标化合物的合成
1.2.1 4-己烷氧基硝基苯(A)的合成
称取4-硝基酚6.95g(0.05mol)放入三口烧瓶中,加入80mL丙酮,再加入8.29g(0.06mol)碳酸钾,加热回流后,加入碘化钾0.33g(0.002mol),再加入等摩尔的溴代正己烷。搅拌回流28小时。反应结束后,抽滤除去无机盐,旋蒸掉大部分溶剂,用乙醚做萃取,用无水硫酸镁干燥,蒸去乙醚,纯化得到黄色液体9.675g,产率86.74%。
1.2.2 4-己烷氧基苯胺(B)的合成
称取二水合氯化亚锡11.28 g(0.05 mol)于250 mL单口圆底烧瓶中,加入1.67mL(0.02mol)浓盐酸,30mL无水乙醇,将0.02mol的化合物A的乙醇溶液用恒压滴液漏斗缓慢滴加到圆底烧瓶中,常温反应1h后,加热回流8h。反应结束后,旋蒸出大部分的乙醇,将混合物倒入烧杯中。在外加冰浴的条件下缓慢加入饱和氢氧化钠溶液,调节pH到9~10时停止滴加。用乙醚萃取有机相,用碱水洗涤至水层无色,再用去离子水洗至中性。乙醚层用氢氧化钠充分干燥,旋蒸除去乙醚。产物在冰箱中放置一夜,得到深褐色固体。用石油醚重结晶得棕白色针状晶体2.92g,产率:75.84%;熔点:42.1~43.5℃。IR ( KBr , v/cm-1):3400,3319,1635(-NH2);3060(AR,C-H);2953~2851(CH3(CH2)5-);1600,1511,1475(Ar)。
1.2.34-((4-(己烷氧基)苯基)偶氮基)苯酚(C)的合成
(1)重氮盐的制备
将提前配制好的稀盐酸(3mol/L)量取40mL加入到250mL的烧杯中,再称取0.04mol4-己烷氧基苯胺加入其中,电磁搅拌使之完全溶解,颜色为藕荷色。然后,用将烧杯置于冰盐浴中,温度控制在0~3℃,此时,由于温度较低,胺盐可能析出,但是不会妨碍下一步的反应。
另称取0.04mol亚硝酸钠溶于20mL蒸馏水,在0~3℃,不断搅拌下,将其慢慢滴入上述烧杯中,滴加完后,继续反应30分钟(0~5℃),反应液的颜色逐渐变为紫红色。反应过程中的体系温度保持在0℃左右,溶液pH值一般保持在3以下。即得所需的重氮盐溶液(保持0~5℃)。应注意的是,此重氮盐不稳定,应立即用于下一步的合成。
(2)4-((4-(己烷氧基)苯基)偶氮基)苯酚的合成
称取0.21mol氢氧化钠溶于20mL蒸馏水。称取0.04mol苯酚,加入盛有氢氧化钠溶液的烧杯中,搅拌使之溶解,加水稀释,将该混合物加入到250ml单口烧瓶中,电磁搅拌,用稀盐酸调体系的pH值在10左右,在冰盐浴中控制温度在0℃左右,一边搅拌,一边慢慢滴加重氮盐溶液,反应过程中有沉淀析出,颜色变化如下:黄—橙黄—砖红。在滴加重氮盐的过程中,若反应体系的pH值下降,可加入适量氢氧化钠溶液来调节pH值,使之一直保持在10左右。重氮盐滴加完毕后,颜色为砖红色,继续搅拌半个小时左右。反应结束后,抽滤,滤饼为橙黄色。将橙黄色固体用冰醋酸重结晶3次,得到橙黄色片状固体。产率56%。熔点93℃。IR (KBr,v/cm-1):3473.76(O-H);2936,2869(CH3(CH2)5-);1599.92(N=N);1575,1498.23,1474.78(Ar);1426(O-H的弯曲振动);1317.79(C-N);1246.50(C-O)。
1.2.44-((4-己烷氧苯基)偶氮基)苯基异烟酸酯(D)的合成
在干燥的烧瓶中加入0.01mol的异烟酸,60mL二氯甲烷。在外加冰浴的条件下加入0.01mol的DCC,反应混合物搅拌10min后,加入适量DMAP和0.01mol的化合物C,常温搅拌72h,在整个反应过程中用TLC跟踪。反应结束后,旋蒸部分溶剂,过滤除去固体,蒸干滤液,用热水将固体洗涤三遍,抽滤,然后用少量乙酸乙酯洗涤两遍,抽滤,旋蒸溶剂,产生黄色固体,再用乙酸乙酯溶解固体,在0℃下静置一夜,然后抽滤,旋蒸溶剂,得到黄色固体,即为化合物D。固体用乙酸乙酯和乙醇(3∶1)的混合溶剂重结晶两次得到黄色片状晶体。产率35%。IR (KBr,v/cm-1):3473.76(O-H);2936,2869(CH3(CH2)5-);1600.36(N=N);1575,1498.23,1474.78(Ar);1426(O-H的弯曲振动);1317.79(C-N);1246.50(C-O)。1H NMR(δ,氘代氯仿,400MHz):8.877(m,2H,ArH);8.034~8.016(m,4H,吡啶环);7.376(m,2H,ArH);7.021(m,2H,ArH);3.045(m,2H,-CH2O-);1.825(m,2H,-CH2-);1.489(m,2H,-CH2-);1.383(m,4H,-CH2-CH2);0.929(m,3H,-CH3)
1.2.5 对烷氧基苯甲酸(E)的合成
在三口圆底烧瓶中,加入1.38g(0.01mol)对羟基苯甲酸,加入50mL乙醇和少量去离子水,搅拌,然后加入1.12g(0.02mol)KOH固体,升温到78℃,然后滴加0.01mol的溴代正己烷和适量KI,回流搅拌,反应24小时。反应结束后,加水稀释,滴加盐酸析出白色沉淀,调节pH值至4,抽滤,滤饼用热水洗涤三次,得到的白色固体用无水乙醇和水(3∶1)的混合溶剂重结晶3次。最终得到白色针状(片状)晶体。产率72%。IR(KBr),v/cm-1:3300~3400(羧-OH);3067(Ar,C-H);2860~2956(CH3(CH2)5-);1672(C=O);1430~1600(Ar);1HNMR(氘代丙酮,400MHz),δ:10.214(s,1H,-COOH);7.882~7.901(m,2H,Ar);7.012~7.067(m,2H,Ar);4.088~4.131(m,2H,-OCH2);2.153~2.162(m,2H,-CH2);1.945~1.990(m,2H,-CH2);1.537~1.621(m,4H,-CH2-);1.105~1.178(m,3H,-CH3)。
1.2.6 超分子化合物(F)的合成
将等摩尔的化合物D和系列化合物E溶于20ml的二氯甲烷中,室温搅拌,直至蒸干溶剂, 真空干燥24h,得到一系列白色超分子化合物F。
2 结果与讨论
2.1 目标化合物的合成
在4-((4-(己烷氧基)苯基)偶氮基)苯酚的合成过程中,影响这个反应的重要因素是反应温度和偶合反应溶液的pH值。在重氮化反应过程中,用冰盐水浴控制体系温度在0~5℃以内,加入适量的亚硝酸钠,保证重氮化反应进行完全。在偶合过程中,pH值的控制是整个反应的重点,pH值gt;11,会产生很多的副反应,而pH值过低的话,又会阻止偶合反应的继续进行,所以在进行偶联反应时,慢慢滴加氢氧化钠溶液使体系的pH保持在9~10。
4-((4-己烷氧苯基)偶氮基)苯基异烟酸酯的合成,采用DCC和DMAP存在下和4-((4-(己烷氧基)苯基)偶氮基)苯酚进行Steglich酯化反应,常温反应3d,副反应较少,后处理简单。
2.2 化合物液晶性能的研究
本实验合成的目标化合物的介晶性通过偏光显微镜对其进行研究。通过在偏光显微镜我们对质子受体 4-((4-己烷氧苯基)偶氮基)苯基异烟酸酯进行液晶性测定,升温至129℃时融解且出现液晶相;继续升温,当升温至168.5℃时,开始出现向列相液晶相;降温过程出现和升温一样的织构,直到108℃时出现结晶,见图2、图3、图4。

图2 化合物D升温132℃的液晶织构

图3 化合物D升温168℃的液晶织构

图4 化合物D降温166℃的液晶织构
对于化合物D和通过分子间氢键形成的偶氮型氢键液晶复合物,其相变温度见表1。

表1 质子受体(D)和氢键超分子化合物(F)的相转变温度(℃)
表1标注:Cr:crystal=晶体; SmX:Smetic=近晶相; N: nematic=向列相; I:isotropic liquid=各向同性液体
氢键复合物与氢键受体相比,其液晶相范围明显拓宽,可能是由于氢键的诱导作用,使得分子间可极化程度增加,诱导偶极可以出现更宽的范围,从而使分子间形成定向排列后更不容易被打乱,使得复合物的的液晶相范围增宽。
在热台偏光显微镜下观察以4-((4-己烷氧苯基)偶氮基)苯基异烟酸酯为氢键受体,4-己烷氧基苯甲酸为氢键供体形成的氢键液晶复合物的液晶织构,这种偶氮型氢键液晶复合物随温度的变化,可以清晰的看到复合物的液晶态的织构。氢键复合物升温到达熔点后,出现了如图5所示织构,继续升高温度,到185℃出现了典型的向列相织构(如图6所示),随着温度的继续升高,到189.5℃转变为各向同性的液体。降温到清亮点时,首先出现如图7所示的球状向列相织构,接着出现一些的弯曲的线状,然后随着温度的下降,这些弯曲的线状能够保持到化合物的另外一个相的形成甚至结晶态。
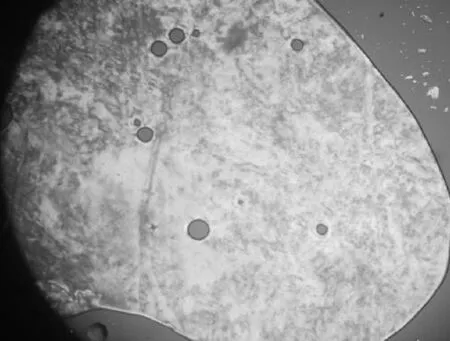
图5 升温163℃液晶织构图

图6 升温189℃液晶织构图

图7 降温185.2℃液晶织构图
3 结论
以4-((4-烷氧苯基)偶氮基)苯基异烟酸酯为氢键受体,4-己烷氧基苯甲酸为质子供体,利用分子间氢键合成了氢键自组装液晶复合物。研究结果表明:氢键液晶复合物的液晶相范围明显拓宽,可能是由于氢键的诱导作用,使得分子间可极化程度增加,诱导偶极可以出现更宽的范围,从而使分子间形成定向排列后更不容易被打乱,致使复合物的液晶相范围增宽。相态及液晶行为研究证明了氢键复合物呈现明显的近晶相和向列相液晶态。超分子液晶复合物较质子供体的液晶相范围要宽很多,说明分子间氢键可以起到稳定液晶相态的作用。
[1] 赵琼,阮班锋,吴杰颖,田玉鹏,等.新型偶氮化合物的合成及其光学性质.合成化学,2009,17(4),450-452.
[2] 阳岑.偶氮苯类液晶化合物的合成、表征及应用研究:(硕士学位论文).兰州:兰州交通大学,2008.
[3] 张会旗,李晨曦,黄文强,等.带双甲基丙烯酰基偶氮苯液晶化合物的合成与表征.化学试剂,1999,21(6),321-323.
[4] 毕思玮.带端烯基偶氮苯液晶化合物的合成和表征.化学试剂,1997,19(5),268-269.
[5]焦家俊.T-型氢键液晶复合物的制备与液晶行为[J].功能高分子学报,2007,2:209-215.
[6]Kato T. Hydrogen-Bonded Liquid Crystals. Novel Mesogens Incorporating Nonmesogenic Bipyridyl Compounds through Complexation between H-Bond Donor and Acceptor Moieti[J].Chem Mater,1993,5:1094-1100.
[7]Kato T.Supramolecular Liquid-Crystalline Complexes Exhibiting Room-Temperature Mesophases and Electrooptic Effects. Hydrogen-Bonded Mesogens Derived from Alkylpyridines and Benzoic Acids[J]. Chem Mater,1995,7:368-372.
[8]Kohmoto Shigeo.Hydrogen-bonded ionic liquid crystals: pyridinylmethylimidazolium as a versatile building block[J]. Tetrahedron Letters,2010,51:1508-1511.
[9]Bruce Duncan W, Halogen-bonded liquid crystals[J]. Struct Bond,2008,126:161-180.
[10]Yagai S, Karatsu T, Kitamura A. Photocontrollable Self-assembly[J ]. Chem Eur J, 2005, 11: 4054-4063.
2017-04-27
王玉辉,男,1963年出生,辽宁石油化工大学本科毕业,在职研究生学历,工程硕士,高级工程师职称,现任神华包头煤化工有限责任公司人力资源部经理,从事石油化工生产及质量检测工作30余年,E-mail:wangyuhui@csclc.com。
信息简讯
超灵敏二硫化钼湿度传感器研究获进展
中国科学报讯现阶段对二硫化钼湿度传感器的研究主要受制于加工过程本身引入的残胶对材料表面的污染,影响了其对水分子的吸附,从而导致灵敏度不高或响应时间过长等问题。因而,如何得到具有高灵敏、快速响应时间的二硫化钼湿度传感器成为制约其应用的最主要因素。
针对上述问题,日前,中国科学院物理研究所/北京凝聚态物理国家实验室(筹)纳米物理与器件实验室利用一种新的金剥离方法,加工得到具有干净表面的二硫化钼场效应晶体管,从而实现了对水分子的灵敏响应。该项工作由实验室博士赵静在研究员张广宇的指导下完成。
据悉,这种加工方法主要是利用二硫化钼与金之间的作用力远大于金与衬底间的作用力,从而可以将多余的二硫化钼样品从衬底上完整地剥离下来,同时保证了用于器件的二硫化钼表面的干净。利用这种方法一方面有效避免了加工过程中经过反应离子刻蚀后表面残胶对器件性能的影响,另一方面大大简化了加工过程, 得到了具有超洁净表面的二硫化钼场效应晶体管,其光学、电学性能的显著提高也从另一个方面证明了这种加工方法得到的样品具有更好的性能。
由于利用这种金剥离方法得到的二硫化钼场效应晶体管具有超洁净的表面,因此能够灵敏感知外界湿度变化,大大提高了二硫化钼湿度传感器的灵敏度。除了具有超高灵敏度外,由于二硫化钼表面没有悬挂键,对水分子的吸附是纯粹的物理吸附,因此器件可以很容易地进行脱吸附,有效缩短了响应时间和恢复时间。除此之外,得益于CVD生长的二硫化钼成膜均匀,可以加工得到一系列具有优异性能的二硫化钼湿度传感器阵列,从而对外界不同湿度的空间分布起到定位作用,用来实时监测外界湿度分布的变化。
这种基于超洁净表面的二硫化钼样品加工得到的湿度传感器具有灵敏度高、响应时间和恢复时间短、使用寿命长、空间分辨率高等特性,可以广泛应用于未来无接触定位系统及二维材料多功能柔性传感器阵列领域。(沈春蕾)
Synthesisandcharacterizationofanovelliquidcrystallinecompound.
WangYuhui,LiuShangqing,ZhaoLingchun,WangTao
(BaotouShenhuaCoalChemicalsCO.,Ltd.,Baotou014010,China)
A new hydrogen bonded supramolecular liquid crystal compound was synthesized.4-((4-(hexoxy) phenyl) azo) phenyl isonicotinic acid ester which played the role of the proton receptor,was synthesized through etherification, reduction, diazotization, coupling reaction and esterification reactions,and 4-alkoxybenzoic acid was as the proton donor.The structures of proton donor and supermolecular liquid crystalline compounds were identified by IR and1H-NMR. Their liquid crystalline phase transitions were studied by optical polarization microscope. The results of optical polarization microscope showed that the supermolecular liquid crystalline compounds were typically thermotropic,which exhibited both the smetic and nematic phases. These supermolecular liquid crystalline compounds were with wide phase transition temperatures. Therefore, it was concluded that liquid crystalline phase could be stabilized by the intermolecular hydrogen bonding action.
azo;smetic liquid crystalline;nematic liquid crystalline
10.3969/j.issn.1001-232x.2017.06.023
猜你喜欢
石油化工(2022年8期)2022-09-07
环境工程技术学报(2022年3期)2022-06-05
陶瓷学报(2021年4期)2021-10-14
成都大学学报(自然科学版)(2021年1期)2021-05-22
石油化工(2021年3期)2021-04-08
表面工程与再制造(2019年1期)2019-05-11
中国粮油学报(2016年1期)2016-02-06
中国铸造装备与技术(2015年5期)2015-12-10
化工进展(2015年6期)2015-11-13
中国洗涤用品工业(2015年4期)2015-02-28
