
胆甾醇基γ-聚谷氨酸负载阿霉素纳米胶束的制备与体内外释药性能评价
2017-12-06 18:22姚俊,肖港,徐宁,曹新,徐虹
生物加工过程 2017年6期
姚 俊,肖 港,徐 宁,曹 新,徐 虹
(1. 南京医科大学 基础医学院 生物技术系,江苏 南京 211166;2. 南京工业大学 食品与轻工学院 材料化学工程国家重点实验室,江苏 南京 211800)
胆甾醇基γ-聚谷氨酸负载阿霉素纳米胶束的制备与体内外释药性能评价
姚 俊1,肖 港1,徐 宁1,曹 新1,徐 虹2
(1. 南京医科大学 基础医学院 生物技术系,江苏 南京 211166;2. 南京工业大学 食品与轻工学院 材料化学工程国家重点实验室,江苏 南京 211800)
笔者制备了胆甾醇基γ-聚谷氨酸负载阿霉素纳米胶束(DOX/NPs),并考察了该载药纳米胶束体系的形态与粒径、载药量、包封率以及体内外释药的特性。结果表明:DOX/NPs的最佳载药量为22.4%,包封率为90.2%,平均粒径为(312.3±7.2) nm,电镜下观察呈现明显的核壳结构。体外释药结果显示,DOX/NPs能延缓阿霉素的释放,并具有pH敏感的释药特性。小鼠体内释药结果表明:阿霉素经包埋后其消除半衰期(t1/2)、药时曲线下面积(AUC)、平均滞留时间(MRT)均明显大于游离阿霉素,达到了药物缓释的目的。
纳米胶束;γ-聚谷氨酸;阿霉素;释药;药代动力学
天然聚氨基酸是目前除多糖外研究较多的一种重要的功能高分子材料,因其具有良好的组织亲和性、非免疫原性和生物可降解性,应用于生物医用材料领域极具优势。γ-聚谷氨酸(γ-polyglutamic acid,γ-PGA)是一种由微生物发酵合成的生物高分子材料,其主链由D,L-谷氨酸单体经γ-酰胺键结合形成Nylon-4结构,在机体内可被降解、吸收、代谢和排泄,不易产生积蓄和毒副作用[1-2],其侧链游离的羧基使之具有优于天然多糖的水溶性和化学改性性能,初步研究已显示出γ-PGA在药物传输领域的应用潜力,特别是采用两亲性嵌段/接枝γ-PGA衍生物以自组装方式获得功能性纳米载药胶束是目前的研究热点[3-6]。本课题组在前期研究中成功构建了两亲性胆甾醇基γ-PGA衍生物(cholesterol-bearingγ-PGA,γ-PGA-graft-CH)及其自组装胶束(γ-PGA-graft-CH NPs)系统[7-8],本研究中,笔者将进一步考察γ-PGA-graft-CH NPs对阿霉素(doxorubicin,DOX)的负载性能以及体内外释药特性,以期为其作为抗肿瘤药物的控缓释载体提供初步参考。
1 材料与方法
1.1 试剂与仪器
γ-PGA-graft-CH NPs(笔者实验室自制,粒径(299.6±5.4) nm,粒径多分散系数为1.2)[8];盐酸阿霉素(doxorubicin hydrochloride,DOX·HCl,浙江海正药业有限公司)。其余试剂均为市售分析纯。
JEM-1010型透射显微镜(日本JEOI公司)、Beckman Coulter N4PLUS型粒径分析仪(美国)、SCIENTZ-IID型超声波匀质仪(宁波新芝生物科技股份有限公司)、LGJ-10D型低温冷冻干燥机(北京四环公司)、高效液相色谱仪(HPLC)/色谱柱(Agilent 1200/Li Chrosphere C18,美国安捷伦公司)、Model 680型酶标仪(美国Bio-Rad公司)。
1.2 γ-PGA-Graft-CH NPs对DOX的负载与表征
两亲性γ-PGA-graft-CH衍生物的合成与γ-PGA-graft-CH NPs的制备参见文献[7-8]。在超声条件下向空白γ-PGA-graft-CH NPs悬液(5 mg/mL)中缓慢滴加1.0 mg/mL DOX·HCl溶液,50 r/min下避光振荡12 h,移至透析袋内,12 h更换去离子水1次,24 h后将透析液过膜超滤(截留分子量1×104),即得负载DOX的γ-PGA-graft-CH NPs(DOX/NPs)溶液,4 ℃保存;透射电镜下观察胶束形态,采用粒径分析仪测定其粒径及粒径分布;通过测量480 nm处的吸光度,对照DOX·HCl标准曲线计算DOX浓度。包封率和载药量的计算见式(1)~(2)。
包封率(EE)=(投药DOX的质量-游离DOX的质量)/投药DOX的质量×100%
(1)
载药量(LC)=载药胶束中DOX的质量/载药胶束总质量×100%
(2)
1.3 体外释药实验
取1.2节制备的DOX/NPs溶液2 mL置于透析袋中,密封后分别放入装有48 mL不同磷酸缓冲液(PBS)(pH 4.0、7.4、9.0)的锥形瓶中,37 ℃、50 r/min条件下振荡孵育,定时取3 mL透析介质(同时补入3 mL新鲜介质),检测DOX的含量,方法同载药量测定,每个样本重复测定3次。体外释药率计算见式(3)。
体外释药率=DOX释放量/DOX总质量×100%
(3)
1.4 体内释药实验
清洁级8周C57BL小鼠(体质量20~25 g),雌雄各半,由南京医科大学实验动物中心提供。将小鼠随机分为2组(每组18只),尾静脉注射给药,DOX剂量均为0.2 mg(以每只小鼠100 g计):对照组采用DOX·HCl注射剂,实验组采用DOX/NPs混悬液。给药后,分别于5、10、20、40、80、160、240和320 min时从每组各取3只老鼠,摘眼球取血约0.5 mL于肝素抗凝管,3 000 r/min离心10 min,取上清血浆4 ℃保存。精确吸取100 μL待测血浆加入1 000 μL甲醇,充分摇匀后,5 000 r/min离心5 min,取上清液,HPLC法测定DOX浓度,色谱条件:流动相为乙腈-水(体积比32∶ 68),检测波长233 nm,流速1 mL/min,进样5 μL。药代动力学数据采用Kinetica v5.0软件进行统计处理。
2 结果与讨论
2.1 DOX/NPs的制备与表征
前期研究表明:γ-PGA-graft-CH衍生物具有两亲性和自组装胶束的能力,采用超声探头法可制备得形态规则、粒径均一且热力学稳定的纳米胶束[8]。载药量对γ-PGA-graft-CH NPs负载DOX性能的影响如图1(a)所示。由图1(a)可知:随载药量增加,γ-PGA-graft-CH NPs的载药量逐渐增加;DOX与载体胶束的药载比小于1/10,包封率处于较高水平(>90%);药载比大于1/10时,载药量虽略有提高,但包封率出现显著下降,说明γ-PGA-graft-CH NPs对DOX的负载已达到饱和。从载药量和包封率综合考虑,1/10为最佳药载比(载药量22.4%,包封率90.2%)。
选取药载比为1/10的DOX/NPs,透射电镜下观察其形态规则,结果如图1(b)所示。由图1(b)可知:胶束界面清晰,粒径较为均一(250~350 nm),且胶束呈明显的核壳结构,可见药物负载主要集中于核区。粒度分析结果显示,DOX/NPs的平均粒径在(312.3±7.2) nm,与电镜测定结果相近,粒径分布多分散系数(PDI)为0.23。
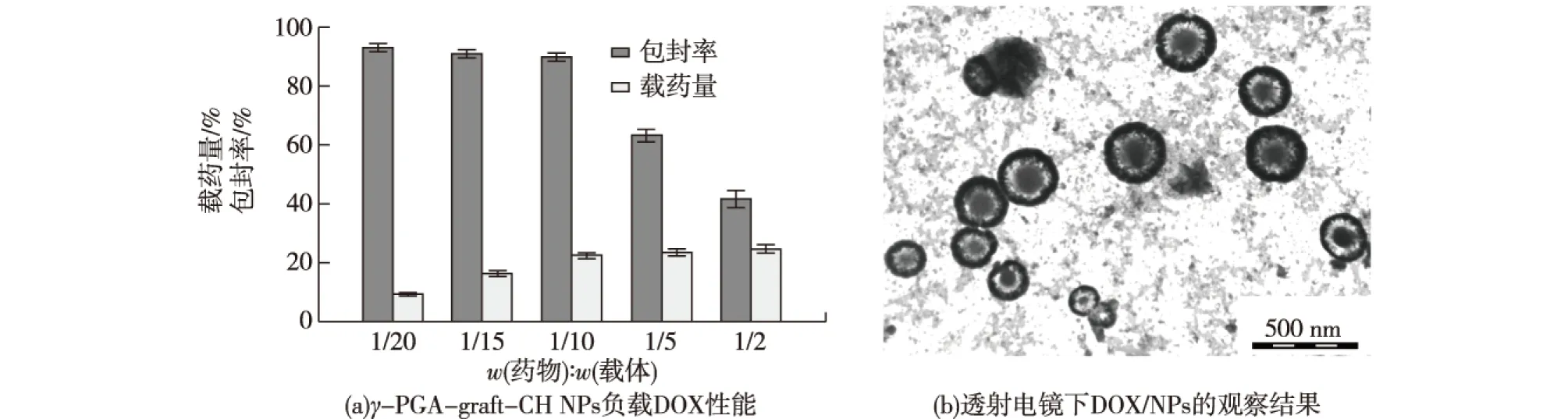
图1 DOX/NPs的制备及性能Fig.1 Preparation and performace of DOX/NPs
2.2 体外释药实验结果
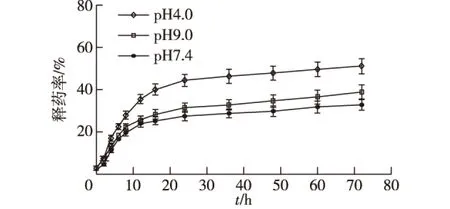
图2 DOX/NPs体外释药曲线Fig.2 Release profiles of DOX from DOX/NPs in vitro
采用不同pH的释药介质考察DOX/NPs的释药性能,结果如图2所示。由图2可知:DOX/NPs在pH 7.4的PBS中释药速率较慢,12 h的释药率仅为23.9%,72 h的释药率为32.9%;在pH 4.0与9.0的PBS介质中,释药速率相对较快,这主要是由于γ-PGA-graft-CH的亲水区域含有大量游离羧基。在偏酸性介质中,γ-PGA-graft-CH亲水段侧链呈质子化趋势,分子内和分子间氢键加强,二级结构以α-螺旋为主,其分子柔性较差,自组装性能以及药物负载能力减弱;在偏碱性介质中,亲水区域因游离羧基电离为—COO-而荷负电,同种电荷相斥使得γ-PGA-graft-CH NPs内部结构吸水溶胀,对包封药物的通透性增加。此外,因药物突释主要与镶嵌或吸附于胶束表面或近表面的药物在短时间内的快速释放有关,而图2显示,DOX经γ-PGA-graft-CH NPs负载后无明显突释现象,说明DOX是包载于γ-PGA-graft-CH NPs核内,与电镜观察结果一致。DOX/NPs释药特性也见于其他pH敏感的凝胶体系中[3-6],由于人体内各组织的环境pH各有差别,特别是肿瘤组织的微环境pH一般低于正常组织。因此,针对这一特性对设计靶向肿瘤组织或特定器官的给药系统具有重要应用价值。
2.3 体内释药实验结果
图3为体内释药实验结果。由图3可知:除给药后的前期(0~5 min),在其余时间点对照组(DOX·HCl)的血药浓度均低于实验组(DOX/NPs);给药后5~40 min,对照组的血药浓度迅速下降,而实验组下降较慢,320 min的血药浓度仍维持在一定水平。此结果表明,DOX经包埋后可有效降低血药峰浓度,并显著延长药物在血液中的滞留时间。
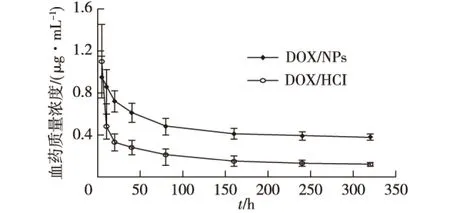
图3 DOX/NPs和DOX·HCl给药后的药时曲线Fig.3 Concentration-time curves of DOX/NPsand DOX·HCl
小鼠血药浓度-时间数据经统计学分析后,主要的药代动力学参数见表1。表1结果显示:对照组小鼠体内DOX代谢极快,其消除半衰期仅66.87 min,这与文献[9]报道的结果(12~100 min)相近;实验组小鼠体内释药缓慢,其消除半衰期(t1/2)、血药浓度-时间曲线下面积(AUC)、平均滞留时间(tMRT)均明显大于对照组(p< 0.01)。此外,实验组的药物清除率(CL)较低,表明DOX/NPs可延长DOX在循环系统内的滞留时间,使得血浆中的药物浓度能维持更长时间,这将有利于改善DOX的生物利用度以及治疗指数。

表1 DOX在小鼠体内的药代动力学参数
注:AUMC—一阶矩曲线下面积;K—消除速率常数;Vss—稳态表观分布体积;Cmax—药峰质量浓度。
3 结论
本研究中,笔者制备的γ-PGA-graft-CH NPs负载DOX的最佳药载比为1/10,对应的载药量为22.4%,包封率达90.2%;获得的载药胶束粒径分布较窄,且DOX主要包埋于γ-PGA-graft-CH NPs的核区。体外释药结果表明:DOX/NPs具有pH敏感的释药特性,且在体内外均显示了良好的缓释性能。综上所述,γ-PGA-graft-CH NPs在抗肿瘤给药领域将极具应用潜力。
[1] 徐虹,欧阳平凯.生物高分子[M].北京:化学工业出版社,2010:18-96.
[2] ASHIUCHI M,MISONO H.Biopolymers[M].Weinheim:Wiley-VCH,2002:123.
[3] FU G D,LI G L,NEOH K G,et al.Hollow polymeric nanostructures:synthesis,morphology and function[J].Prog Polym Sci,2011,36:127-167.
[4] SHIDHAYE S.Advances in polymeric micelles for drug delivery and tumor targeting[J].Nanomed Nanotechnol Biol Med,2010,6:714-729.
[5] RAO J P,GECKELER K E.Polymer nanoparticles:preparation techniques and size-control parameters[J].Prog Polym Sci,2011,36:887-913.
[6] 王龙海,洪春雁.响应性枝化聚合物的合成、组装及其生物应用[J].高分子学报,2017(2):200-213.
[7] 陈宽婷,姚俊,阮文辉,等.新型γ-聚谷氨酸自组装纳米胶束的制备及用于蛋白载体的研究[J].中国生物工程杂志,2013,33(4):101-105.
[8] 李睿,阮文辉,姚俊,等.γ-聚谷氨酸胆甾醇基衍生物自组装纳米胶束的制备与表征[J].高分子材料科学与工程,2014,30(1):11-14.
[9] HAN H D,LEE A,SONG C K,et al.In vivo distribution and antitumor activity of heparin-stabilized doxorubicin-loaded liposome[J].Int J Pharma,2006,313(1/2):181-188.
(责任编辑 管珺)
Preparationofdoxorubicin-loadednanoparticlesofcholesterol-bearingγ-polyglutamicacidanditsdrug-releaseinvivoandinvitro
YAO Jun1,XIAO Gang1,XU Ning1,CAO Xin1,XU Hong2
(1. Department of Biotechnology,School of Basic Medical Science,Nanjing Medical University,Nanjing 211166,China;2. State Key Laboratory of Materials-Oriented Chemical Engineering,College of Food Science and Light Industry,Nanjing Tech University,Nanjing 211800,China)
We prepared adoxorubicin (DOX) loaded nanoparticles of cholesterol-bearingγ-polyglutamic acid (DOX/NPs) and studied the properties of DOX/NPs,including drug loading,drug entrapment efficiency,drug releaseinvitroandinvivo.The optimum loading was 22.4% and entrapment efficiency was 90.2%.The mean size of DOX/NPs was (312.3±7.2) nm with narrow distribution,and a typical core-shell structure was observed via transmission electron microscope.A pH-sensitive sustainable drug-release was detectedinvitro,andinvitroexperiments confirmed the slow-releasing characteristic of DOX/NPs.Compared with DOX·HCl injections,DOX/NPs could significantly enhance the elimination half-life(t1/2),area under concentration-time curveand mean residence time to achieve the slow-release of loaded DOX.
nanoparticles;γ-polyglutamic acid;doxorubicin; drug-release; pharmacokinetics
10.3969/j.issn.1672-3678.2017.06.009
2017-07-02
国家自然科学基金青年基金(51403103);江苏省高等学校大学生实践创新训练计划(201710312065X)
姚 俊(1979—),江苏扬州人,博士,讲师,研究方向:生物医用材料;徐 虹(联系人),教授,E-mail:xuh@njtech.edu.cn
O636.9;R944.1+5
A
1672-3678(2017)06-0064-04
猜你喜欢
中成药(2018年9期)2018-10-09
中成药(2017年5期)2017-06-13
中成药(2017年6期)2017-06-13
中国洗涤用品工业(2017年2期)2017-04-16
中国比较医学杂志(2017年5期)2017-01-17
山西医科大学学报(2016年2期)2016-05-25
医学研究杂志(2015年12期)2015-06-10
医学研究杂志(2015年5期)2015-06-10
中国药理学通报(2014年2期)2014-05-09
中国药理学通报(2014年2期)2014-05-09
