
跨平台的质谱蛋白回归定量和质量控制的参数方法
2017-11-28 05:26刘万霖
质谱学报 2017年6期
魏 来,刘万霖,丁 琛,2,秦 钧
(1.蛋白质组学国家重点实验室,北京蛋白质组研究中心,军事医学科学院放射与辐射医学研究所,北京 102206;2.复旦大学生命科学学院,上海 200032)
跨平台的质谱蛋白回归定量和质量控制的参数方法
魏 来1,刘万霖1,丁 琛1,2,秦 钧1
(1.蛋白质组学国家重点实验室,北京蛋白质组研究中心,军事医学科学院放射与辐射医学研究所,北京 102206;2.复旦大学生命科学学院,上海 200032)
各质谱厂商通常使用不同的软件进行蛋白鉴定和定量,导致获得的数据和结果的通用性不佳。另外,目前蛋白质定量的准确性仍有提升空间。因此,开发一个标准化、自动化且定量更准确的蛋白质定量流程具有现实意义。采用肽段保留时间对齐和回归技术,可有效地减少中低丰度肽段鉴定信息缺失带来的影响,提高中低丰度蛋白的定量能力。一个综合考虑信号峰宽分布、保留时间分布以及肽段同位素模式分布等因素的肽段筛选器,可以有效地过滤掉不适宜定量的肽段信号,使肽段离子流色谱(XIC)峰定量面积的计算更为准确。该流程由数据开源转换、保留时间对齐与回归定量、肽段筛选器等模块构成,可准确定量不同平台产出的质谱数据,并明显改善低丰度蛋白的无标定量。经对比,该流程对蛋白质组学动态范围标准蛋白集(UPS2)的定量比MaxQuant和Proteome Discoverer的定量更准确。
质谱;蛋白定量;离子流色谱(XIC);保留时间
蛋白质组(proteome)是指某一生物体、组织、器官或者细胞,在特定条件下表达的全套蛋白质[1]。蛋白质组学是蛋白质的大规模研究,其目的在于完整地定量描述不同条件下蛋白质的表达及其变化[2-8],最终描绘出人体及其他生命体的蛋白质组全表达谱。
各种生命体表达的蛋白种类繁多且数量庞大,且许多重要蛋白丰度很低,较难检测。故蛋白质组的研究工具应具备高通量和高灵敏度特性。串联质谱的出现实现了蛋白的高通量鉴定乃至定量[9-11]。
质谱技术发展迅速,新一代质谱在分辨率、质量准确性、灵敏度和动态范围等方面均有很大提升[12]。比如Orbitrap Velos的图谱利用率可达50%以上[13],已远超过TOF 20%~30%的图谱利用率。但是,其与硬件配套的软件还不够完善,存在许多问题。首先是软件环境封闭的问题,以常用的免费定量软件MaxQuant为例,它内嵌Andromeda引擎[14]且与之绑定,无法调用Mascot和SeQuest等其他常用引擎的搜库结果。由于常规的质控研究直接针对这些常用的引擎,因而人们难以对MaxQuant的结果进行质量控制,而质量控制对于保证鉴定有效性以及鉴定和定量列表的一致性非常重要[15-17]。其次,部分软件定量的准确性不高,以SIEVE[18]和PinPoint[19]为例,经过人工判读对比,其鉴定和定量结果与实际存在较大差异,且往往存在假峰,定量结果的可信度无法让人满意。由于大部分已有软件存在硬性限制,最新的蛋白质组学研究往往停留在谱图计数水平上,如2014年Nature报道的人类蛋白质组图谱的开创性工作[2]受限于定量精度和定量软件对海量数据处理的可操作性,仍然使用谱图计数方法定量蛋白,对研究的深度而言仍可谓是美中不足。
目前,蛋白定量的有待改进之处是由肽段鉴定信息缺失导致无法充分利用谱图数据中的定量信息。蛋白质鉴定是蛋白质定量的前提[10],通过搜库将质谱谱图与氨基酸序列关联匹配是串联质谱鉴定蛋白的基本原理之一[20]。得到蛋白鉴定信息列表后,可据此从原始谱图根据离子流色谱(XIC)峰强度或面积得到肽段的定量值,并间接得到蛋白的定量值[21-22]。但是,在质谱检测中,由于质谱数据的谱图利用率往往低于50%,因此来源于低丰度蛋白的肽段信号容易被信号更强的肽段掩盖,从而被搜索引擎遗漏,无法获得鉴定信息,更无法得到定量数值。
由于这些蛋白表达量上的变化往往具有重要的生物学指导意义,因此需要对它们进行准确定量。要准确定量这些蛋白,需要找出这些蛋白在鉴定中被遗漏的肽段,可以作为重要依据的是肽段的色谱保留时间。色谱保留时间是指从溶质注射到溶质洗脱出现最大峰值所经过的时间,溶质在色谱中的保留时间由色谱过程中的热力学性质决定。任何一种溶质,在特定色谱条件下,都可以确定其保留时间,保留时间不同则溶质不同。
此外,为了提高蛋白定量的准确性,有必要对肽段进行过滤和筛选。
本方法旨在开发一种标准化、自动化的蛋白定量流程,灵活、充分和准确地利用各主流质谱平台上高分辨率质谱数据。通过色谱保留时间充分挖掘图谱数据中的定量信息,同时基于肽段的多种特征参数及天然同位素分布模式,对肽段进行过滤筛选,从而得到更准确的定量结果,特别是提高中低丰度蛋白定量效率和定量准确度。
1 方法和策略
1.1质谱产出数据的格式转换
质谱数据的格式转换包括:1) 使用ReAdW结合Xcalibur将一级质谱(MS1)数据转换成mzXML开源格式,使其容易解析和使用,转换成mzXML后的图谱数据采用Base64编码,使用JAVA包axis.jar解码;2) 使用Proteome Discoverer调用Mascot进行搜库,并将搜库结果导出为csv文本文件,使鉴定信息容易获取和使用,该软件工作流程示于图1。
流程的主要步骤包括:原始质谱数据转换成开源格式、搜库鉴定结果导出成文本文件、以及使用两者进行回归定量。回归定量的具体步骤包括:从串联质谱鉴定结果中提取肽段信息,利用肽段信息从一级图谱中提取离子流色谱峰信息,对离子流色谱峰做平滑化处理以及计算平滑后的峰面积等。
1.1.1Thermo Scientific质谱平台产出数据的格式转换 以LTQ-Orbitrap Velos为例,首先使用ReAdW结合Xcalibur将raw格式质谱数据转换成mzXML格式。然后使用Proteome Discoverer读取msf格式搜库结果并导出为csv文本文件。确保mzXML文件和csv文件除后缀外的文件名相同。
1.1.2AB Sciex质谱平台产出数据的格式转 换 以TripleTOF 5600为例,首先将wiff格式质谱数据转换成mzXML格式。然后使用MSConvert软件将wiff格式质谱数据转换成mgf格式,在Proteome Discoverer中调用Mascot对mgf文件进行搜库,并将搜库结果导出为csv文本文件。确保mzXML文件和csv文件除后缀外的文件名相同。

图1 软件工作流程Fig.1 Flowchart of approach
1.2提取肽段鉴定信息列表
以LTQ-Orbitrap Velos为例,其产出的raw格式质谱数据可以通过Thermo Proteome Discoverer进行搜库,搜库结果包括:肽段序列、肽段修饰、带电荷数、质荷比和保留时间等信息,从搜库结果导出的csv文件中可以提取一张高置信度肽段列表。
1.3提取离子流色谱峰及定量
首先,从搜库结果列表中提取候选肽段信息,包括蛋白ID、肽段序列、肽段修饰、带电荷数、质荷比、保留时间和扫描数等。如果出现肽段序列、修饰、带电荷数完全相同的多条数据,实际上属于同一个肽段匹配图谱(peptide spectrum match, PSM),源于同一个XIC,因此只需要保留其中tR处于中位数的一条信息作为其代表信息即可。
然后,确定不同实验间的共鉴定肽段集合,对共鉴定肽段集的两组保留时间做回归得到回归方程。根据回归方程估计保留时间,并在保留时间附近的一级谱图中提取出与该候选肽段质荷比相同的所有信号强度值(相同是指质荷比的差在一定误差阈值范围内),将其沿时间轴排列成肽段的离子流色谱峰。对得到的离子流色谱峰做平滑化处理后,使用梯形数值积分计算峰面积作为肽段的定量值。
2 结果与讨论
2.1保留时间回归方程
两次质谱运行的搜库结果之间有一个共鉴定肽段集合,该集合中的肽段在两次搜库结果中都有保留时间,可以通过拟合得到两组保留时间之间的回归方程,并根据一组保留时间和回归方程,预测另一组的保留时间。通过计算保留时间预测值与实际值之间的线性相关系数的平方(R2),可以评估该回归方程预测保留时间的准确程度。在216组成对实验中,线性回归方程的预测值与实际值之间的R2是0.996±0.007(范围区间[0.945, 0.999]),2次多项式回归得到的R2是0.998±0.005(范围区间[0.978, 0.999])。线性回归方程和2次多项式回归方程做出的预测均与实际值基本符合,只是在线性回归拟合曲线的首尾处有轻微位移。
2.2离子流色谱筛选和整合
肽段在色谱中连续分布,但一级质谱扫描时间点是离散的,所以首先要将离散信号点平滑成信号曲线构造离子流色谱峰。多项式滤波器是常用的平滑方法,它通过多项式函数分段地拟合信号。本实验使用Savitzky-Golay滤波器[23]对离散信号点进行平滑化处理,然后用梯形数值积分[24]计算离子流色谱峰的覆盖面积。
2.3利用贝叶斯模型的多参数质量控制
通过设定峰宽、半高峰宽、tR、m/z、峰面积以及Ion Score等参数的阈值对定量结果进行质量控制,通过它们的组合筛选排除掉定量准确度较低的肽段,从而提升定量结果的准确度。实验得到了不同峰宽和半高峰宽的肽段定量准确度差异分布,示于图2。

图2 不同峰宽和半高峰宽的肽段定量准确度差异分布Fig.2 Distribution of variation of quantifications of peptides with different peak width and full width at half maximum
由图2可见,半高峰宽过大的肽段定量准确度差异值较大,表示肽段定量准确性不佳,应当排除以避免影响蛋白定量的准确度。
2.3.1联合似然比 当参数两两相关时,计算参数对的联合似然比不仅可以避免线性独立性的证明,还可以提升方法的准确度。以峰宽和半峰宽为例,其联合似然比示于图3。图中,红色代表似然比高,即真阳性率高;蓝色代表似然比低,即真阳性率低。半高峰宽较高的区域为深蓝色,表示参数值落在此区域的肽段具有较低阳性率。本实验结果表明,XIC的半峰宽较高往往意味着有噪声干扰或信号重叠等现象发生,因此峰的准确度会受到影响。半高峰宽较低及峰宽较高的区域主要是红色,这意味着参数位于该区域的肽段倾向于拥有较高的准确度,这种峰通常干扰较少、峰型较好。相较于单独使用峰宽或者半峰宽,计算峰宽和半峰宽的联合似然比具有更准确的判断力,且可以避免峰宽与半峰宽之间线性独立性证明。
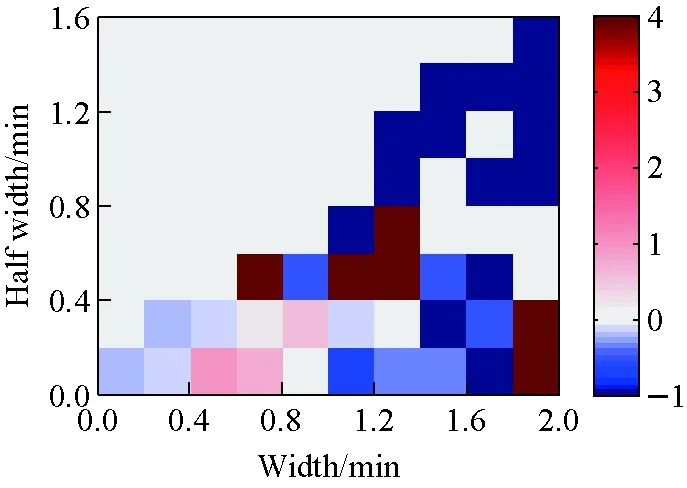
图3 峰宽与半高峰宽的联合似然比Fig.3 Joint likelihood ratio of peak width and full width at half maximum
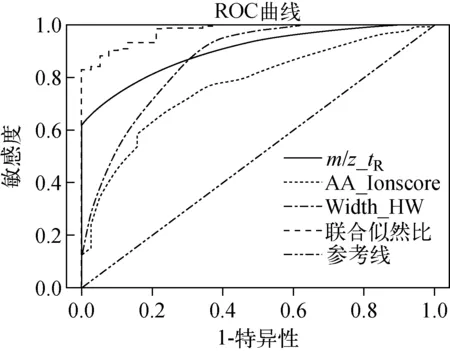
图4 各种参数组合的ROC曲线Fig.4 ROC curves of parameters groups
2.3.2贝叶斯模型的整合 各种参数组合的受试者工作特征曲线(receiver operating characteristic curve, ROC)示于图4。ROC曲线上的每个点代表着检验依据某一似然比卡值预测得到的真阳性率(true positive rate, TPR, Y轴)和假阳性率(false positive rate, FPR, X轴)。ROC曲线下方面积(AUC)是预测模型有效性的量化指标。一个敏感度和特异度均为100%的理想检验的AUC等于1;而一个无效预测模型的AUC等于0.5。一个检验规则的AUC值越接近于1(或接近于0,此时它作为反向指标),预测的总体有效度越高。从图4中可以看出,在m/z+tR、AA+Ion Score、峰宽+半峰宽以及联合似然比等组合的ROC曲线下方覆盖面积(AUC)比较中,联合似然比ROC曲线的AUC值最高,达0.976,表明本实验改进的贝叶斯模型AUC接近于1(0.976),该模型能更好地区分定量结果和异常值,具有最佳准确性。
2.4运用同位素模式进行过滤与校正
自然界中同位素的分布是大体恒定的,这一特点在质谱检测中反映为同一肽段序列存在多种同位素模式,且量的分布服从某种规律。将20 000条肽段数据输入理论模型(IPC程序)后,得到了它们的各种同位素模式的量的理论分布。据此为1~6电荷分别做出每对同位素模式的理论比值与质荷比之间的X-Y坐标关系图。发现nth同位素模式与1st同位素模式的比值与m/z之间存在线性或2次多项式函数关系,因此从回归函数可以得到nth同位素模式与1st同位素模式的理论比值,从而筛除定量不准确的肽段。2电荷肽段和4电荷肽段的结果示于图5,其他电荷数的结果与之相似。
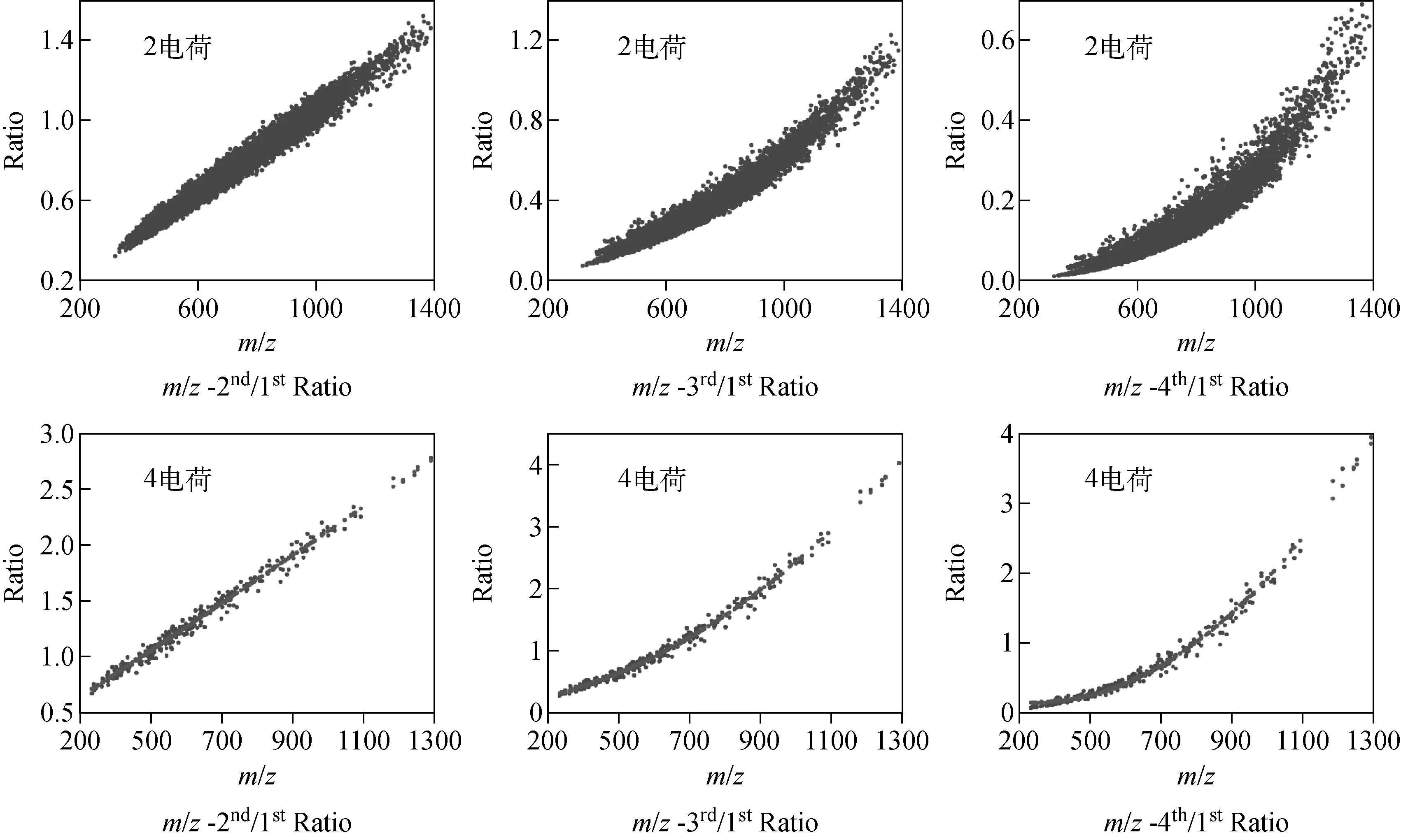
图5 质荷比与同位素模式比值间的拟合Fig.5 Fitting of m/z and the ratio of different isotopic patterns
考虑每个肽段的前3个同位素模式,基于质谱的无标定量,其准确度的相对标准偏差(RSD)为10%~30%[25]。因此,如果第2、第3同位素模式与第1同位素模式的定量值比值超出理论比值±30%,则可以排除系统固有的影响,并认为第1同位素模式的定量值不准确,可将之除去。
通过比较技术重复实验的相关性,证实了在肽段水平上除去定量值位于同位素模式分布理论范围之外的、表现不佳的肽段是一种正确的方式。但如果在一次质谱运行结果中除去表现较差的肽段,而在另一次质谱运行结果中保留它们,将人为扩大蛋白水平上的差异。实验表明,在所有参与比较的质谱运行结果中去除差肽段后,蛋白定量得到了更好的结果。本实验比较了两次技术重复实验数据,进行同位素模式峰过滤后的实验相关性较之未过滤前提高了2~5个百分点,且对不同的仪器R2均有提高。Oribtrap Velos过滤前后的相关性分别为0.954,0.973;QE过滤前后2次实验的相关性分别为0.879,0.936;而QE plus的结果分别为0.947 和0.970。
2.5与其他方法的比较
蛋白质组学动态范围标准样品(proteomics dynamic range standard, or universal proteomics standard 2, UPS2)包括含量分布在6个数量级中,48种已知浓度的标准蛋白,是检测定量方法低丰度覆盖率和定量准确度的常用样品。将UPS2标准品稀释10倍后,用本方法能够检测出各个数量级丰度下的蛋白,特别是低丰度蛋白的准确定量值,其定量值与蛋白实际丰度之间的相关度R2显著高于Proteome Discoverer和MaxQuant方法,且可定量的蛋白数量均高于另外两种方法,详细结果列于表1。这一结论对标准品稀释100倍及1 000倍的情况同样成立。

表1 不同方法测得标准蛋白的R2和定量蛋白数Table 1 R2 and number of proteins using different methods on UPS2
注:iBAQ: Intensity based absolute quantification,基于强度的绝对定量;LFQ: Label free quantification,MaxQuant软件的无标定量;#pro表示该方法在该浓度下可得到鉴定值的蛋白数量
2.6应用与验证
在昼夜节律研究中,将本方法得到的鼠肝中的蛋白质表达量的昼夜节律变化的蛋白质组定量结果与先前包含WAT、BAT、Liver、Muscle的转录组时间节律研究中[26]已公开的转录组数据中肝脏中相关基因表达丰度进行比较,两者变化高度一致,呈相关趋势。通过tumor necrosis factor-α (TNFα)刺激293T细胞的实验中,利用TNFα激活NF-κB信号通路,然后用人工合成的sdTF反应元件pull-down样品293T核抽提蛋白,通过检测转录因子的变化,全景式描述NF-κB信号通路。利用本实验的定量策略分析质谱检测结果,显示受TNFα刺激后,NF-κB家族中NFκB1和RelA有显著上调,这与文献报道结果一致[27],且相关通路中其他转录因子变化均符合文献报道。通过定量结果分析,能够更好地解释TNFα激活NF-κB信号通路后细胞中转录因子的动态变化,以及阐释不同蛋白之间的相互作用。综上可见,本方法得出的结果可准确反映生物学现象。
3 结论
基于高精度质谱数据开发了自动化、标准化的定量流程,该流程具有良好的通用性,可用于包括Thermo Fisher和AB Sciex等主流高分辨质谱在内的多种质谱平台。该流程产出的数据采用开源标准格式,有利于中间数据的再利用,同时流程本身的松耦合、模块化设计便于对结果进行质量控制或以之为组件构建新工作流。本实验开发的保留时间回归和对齐技术能够充分利用鉴定信息挖掘谱图中隐藏的定量信息,有效地改善中低丰度蛋白的定量覆盖度和准确度,对具有重要生物学研究价值的低丰度蛋白定量意义重大。此外,还探讨了肽段的各特征参数对其定量准确度的影响,并利用联合似然方法将多种特征参数加以整合,能够更为准确地筛选出定量准确的肽段,使蛋白的定量准确度得到进一步改善。实验中还考虑了肽段同位素模式的分布规律,利用肽段同位素模式的理论分布模型,筛除了没有准确定量的肽段。
基于蛋白质组学动态范围标准蛋白UPS2比较了本方法与MaxQuant、Proteome Discoverer的定量性能,结果表明,本方法更为准确。在昼夜节律研究中,本实验的方法定量结果与文献报道一致,可准确反映生物学现象。本实验中开发的自动化、标准化定量流程使蛋白质的无标定量变得更加准确、便捷和通用,作为定量蛋白质组学乃至生物学研究的基础环节,新的、更为准确的蛋白质质谱定量方法将为蛋白质的生物学研究和临床应用打下更为坚实的数据基础。
致谢:感谢国防科学技术大学张伟博士在定量工作中的指导和帮助。
[1] PANDEY A, MANN M. Proteomics to study genes and genomes[J]. Nature, 2000, 405(6 788): 837-846.
[2] KIM M S, PINTO S M, GETNET D, et al. A draft map of the human proteome[J]. Nature, 2014, 509(7 502): 575-581.
[3] WILHELM M, SCHLEGL J, HAHNE H, et al. Mass-spectrometry-based draft of the human proteome[J]. Nature, 2014, 509(7 502): 582-587.
[4] BROWER V. Proteomics: biology in the post-genomic era[J]. EMBO Reports, 2001, 2(7): 558-560.
[5] OMENN G S, STATES D J, ADAMSKI M, et al. Overview of the HUPO plasma proteome project: results from the pilot phase with 35 collaborating laboratories and multiple analytical groups, generating a core dataset of 3020 proteins and a publicly-available database[J]. Proteomics, 2005, 5(13): 3 226-3 245.
[6] YING W, JIANG Y, GUO L, et al. A dataset of human fetal liver proteome identified by subcellular fractionation and multiple protein separation and identification technology[J]. Molecular amp; Cellular Proteomics, 2006, 5(9): 1 703-1 707.
[7] HAMACHER M, APWEILER R, ARNOLD G, et al. HUPO brain proteome project: summary of the pilot phase and introduction of a comprehensive data reprocessing strategy[J]. Proteomics, 2006, 6(18): 4 890-4 898.
[8] JIANG Y, YING W, WU S, et al. First insight into the human liver proteome from PROTEOMESKY-LIVERHu 1.0, a publicly available database[J]. J Proteome Res, 2010, 9(1): 79-94.
[9] MARCOTTE E M. How do shotgun proteomics algorithms identify proteins?[J]. Nature Biotechnology, 2007, 25(7): 755-757.
[10] AEBERSOLD R, MANN M. Mass spectrometry-based proteomics[J]. Nature, 2003, 422(6 928): 198-207.
[11] TOLMACHEV A V, MONROE M E, PURVINE S O, et al. Characterization of strategies for obtaining confident identifications in bottom-up proteomics measurements using hybrid FTMS instruments[J]. Analytical Chemistry, 2008, 80(22): 8 514-8 525.
[12] GEIGER T, COX J, MANN M. Proteomics on an Orbitrap benchtop mass spectrometer using all-ion fragmentation[J]. Molecular amp; Cellular Proteomics, 2010, 9(10): 2 252-2 261.
[13] DEUTSCH E W, LAM H, AEBERSOLD R. Data analysis and bioinformatics tools for tandem mass spectrometry in proteomics[J]. Physiological Genomics, 2008, 33(1): 18-25.
[14] COX J, NEUHAUSER N, MICHALSKI A, et al. Andromeda: a peptide search engine integrated into the MaxQuant environment[J]. Journal of Proteome Research, 2011, 10(4): 1 794-1 805.
[15] CARR S, AEBERSOLD R, BALDWIN M, et al. The need for guidelines in publication of peptide and protein identification data working group on publication guidelines for peptide and protein identification data[J]. Molecular amp; Cellular Proteomics, 2004, 3(6): 531-533.
[16] NESVIZHSKII A I, AEBERSOLD R. Analysis, statistical validation and dissemination of large-scale proteomics datasets generated by tandem MS[J]. Drug Discovery Today, 2004, 9(4): 173-181.
[17] CHAMRAD D, MEYER H E. Valid data from large-scale proteomics studies[J]. Nature Methods, 2005, 2(9): 647-648.
[18] DUNN W B, BROADHURST D, BROWN M, et al. Metabolic profiling of serum using ultra performance liquid chromatography and the LTQ-Orbitrap mass spectrometry system[J]. Journal of Chromatography B, 2008, 871(2): 288-298.
[19] CAMPBELL J, REZAI T, PRAKASH A, et al. Evaluation of absolute peptide quantitation strategies using selected reaction monitoring[J]. Proteomics, 2011, 11(6): 1 148-1 152.
[20] LU B, CHEN T. Algorithms for de novo peptide sequencing using tandem mass spectrometry[J]. Drug Discovery Today: BioSilico, 2004, 2(2): 85-90.
[21] GRIFFIN N M, YU J, LONG F, et al. Label-free, normalized quantification of complex mass spectrometry data for proteomic analysis[J]. Nature Biotechnology, 2010, 28(1): 83-89.
[23] SAVITZKY A, GOLAY M J E. Smoothing and differentiation of data by simplified least squares procedures[J]. Analytical Chemistry, 1964, 36(8): 1 627-1 639.
[24] WEIDEMAN J A C. Numerical integration of periodic functions: a few examples[J]. The American Mathematical Monthly, 2002, 109(1): 21-36.
[25] XIE F, LIU T, QIAN W J, et al. Liquid chromatography-mass spectrometry-based quantitative proteomics[J]. Journal of Biological Chemistry, 2011, 286(29): 25 443-25 449.
[26] YANG X, DOWNES M, RUTH T Y, et al. Nuclear receptor expression links the circadian clock to metabolism[J]. Cell, 2006, 126(4): 801-810.
[27] PERKINS N D. Integrating cell-signalling pathways with NF-κ B and IKK function[J]. Nat Rev Mol Cell Biol, 2007, 8(1): 49-62.
ParametricApproachforRegressionQuantificationandQualityControlofProteinsfromMultipleMassSpectrometryPlatforms
WEI Lai1, LIU Wan-lin1, DING Chen1,2, QIN Jun1
(1.StateKeyLaboratoryofProteomics,BeijingProteomeResearchCenter,BeijingInstituteofRadiationMedicine,NationalCenterforProteinSciences(ThePHOENIXCenter,Beijing),Beijing102206,China; 2.StateKeyLaboratoryofGeneticEngineeringandCollaborativeInnovationCenterforGeneticsandDevelopment,SchoolofLifeSciences,InstitutesofBiomedicalSciences,FudanUniversity,Shanghai200032,China)
Quantitative analysis based on high-resolution mass spectrometry data, as an increasingly popular high-throughput method in the past decade, is a fundamental work in proteomic research. Different MS manufactures typically use different suite of software for protein identification and quantification. It is inconvenient to compare results from different MS platforms. Furthermore, the accuracy of protein quantification is improvable. Hence, the development of a standardized and automated protein quantification process is urgent for proteomics researches. A parametric approach was established for quantitative research based on mass spectrum. First, basic information of candidate peptides was extracted from mainstream spectra search program, mainly includem/z, retention times (tR), and corresponding protein IDs, peptides, modifications, charge states, as well as scan numbers. Then the intensities of peptides in first level in tandem mass spectrometry (MS1) were retrieved from the decoded raw file basing onm/zandtRin a proper threshold. The extracted intensity values around the retention time for a certain peptide can be constructed into extracted ion chromatograms (XIC) peak. After smoothed by Savitzky-Golay smoothing filter, the XIC peak area was calculated by trapezoidal rule. For the peptides produced MS1signals but not MS2signals due to random sampling in MS, regression was used to improve parallelism of pairwise experiments. Polynomial curve fitting of degree 2 was used to retrieve regression function of retention time by common identified peptides between different runs. With the retention time alignment and regression approach, quantification for low abundance proteins can be improved greatly. A parametric model based on the peak width, retention time and other parameters were combined by joint likelihood ratio and constructed by Bayesian model. This approach can effectively filter out poor peptide for quantification. By evaluated the relationship ofm/zand the ratio ofnth/1stisotope pattern intensities based on theory isotope pattern models, the function relationships were determined betweenm/zand the ratio ofnth/1stisotope pattern intensities, and the functions for every charge state were built. Then their theory intensities of isotope patterns can be calculated simply. And discarding the poor peptides that out of the theory isotope pattern range based on isotope pattern is an effective way to improve the accuracy of quantification. A comprehensive filter with all these factors can adjust the quantification of XIC. This standardized and automated process contains modules for data format conversion, retention time alignment and regression, and a multi-parametric peptide filter. It works on the majority of mainstream high resolution tandem mass spectrum and derived more precise quantification results, especially for low abundance proteins. A comparison among this process, MaxQuant and Proteome Discoverer proved the accuracy of this parametric method for protein quantification. The method provides a more accurate and intuitive method for MS data utilization, a more convenient method for protein quantification researches.
mass spectrum; protein quantification; extracted ion chromatogram (XIC); retention time (tR)
O657.63
A
1004-2997(2017)06-0611-09
10.7538/zpxb.2017.0010
2017-01-16;
2017-03-28
国际科技合作项目《蛋白质组信息学关键技术及分析系统研发》(2014DFB30010);青年科学基金项目《利用高解析度质谱数据进行中低丰度蛋白质无标定量的研究》(31200992)资助
魏 来(1989—),男(汉族),辽宁建平人,硕士研究生,生物化学与分子生物学专业。E-mail: wlai_bprc@126.com
丁 琛(1982—),男(汉族),安徽滁州人,研究员,从事蛋白质组学研究。E-mail: chend@fudan.edu.cn
秦 钧(1965—),男(汉族),辽宁大连人,研究员,从事蛋白质组学研究。E-mail: jqin@bcm.edu
猜你喜欢
科海故事博览·下旬刊(2022年4期)2022-05-07
宇航计测技术(2021年1期)2021-08-17
江苏农业科学(2021年1期)2021-03-15
科学技术创新(2021年2期)2021-01-21
同位素(2018年1期)2018-01-18
价值工程(2016年32期)2016-12-20
电子制作(2016年24期)2016-04-18
中小企业管理与科技·中旬刊(2014年10期)2015-02-03
同位素(2014年3期)2014-06-13
