
常规食用调味面制品对青年志愿者肠道菌群多样性影响
2017-11-21 10:50丹丹
食品工业科技 2017年21期
, , ,,丹丹,, ,*
(1.湖北文理学院化学工程与食品科学学院 鄂西北传统发酵食品研究所,湖北襄阳 441053; 2.当阳市妇幼保健院检验科,湖北宜昌 444100; 3.襄阳出入境检验检疫局综合实验室,湖北襄阳 441003)
常规食用调味面制品对青年志愿者肠道菌群多样性影响
蔡宏宇1,王艳2,沈馨1,马雪伟3,王丹丹1,余海忠1,郭壮1,*
(1.湖北文理学院化学工程与食品科学学院 鄂西北传统发酵食品研究所,湖北襄阳 441053; 2.当阳市妇幼保健院检验科,湖北宜昌 444100; 3.襄阳出入境检验检疫局综合实验室,湖北襄阳 441003)
招募了9名在校大学生志愿者,每位志愿者每隔一天食用一包约150 g的调味面制品,持续2周累计食用7包,分别于食用前一日、食用完次日及2周后采集粪便样品,并采用高通量测序技术对其肠道微生物群落结构进行解析。通过配对t检验发现,食用调味面制品2周可以显著提高志愿者肠道微生物的多样性和丰富度(p<0.05),而对优势菌属的相对含量没有影响(p>0.05)。通过基于UniFrac距离的主坐标分析和基于Meta-Storm距离的非参数多元方差分析发现,食用调味面制品2周不会对志愿者肠道菌群群落结构产生影响。
调味面制品,高通量测序,肠道微生物,青年
调味面制品,是以小麦粉为主要原料,经配料、挤压熟制、成型、调味而成的即食食品[1-2],该类产品鲜、咸、香、辣、油,口感十分丰富,深受青少年儿童的青睐,2015年销售额达到500亿[3]。调味面制品的生产包含油浸和调味等工艺环节,具有高油、高盐和辛辣的特点,且含有色素、甜味剂和防腐剂等食品添加剂。有报道指出调味面制品市场抽检合格率一般在80%左右[3],加之质量安全问题偶有发生,因而在一定程度上引发了消费者的担忧[4]。然而令人遗憾的是,目前关于调味面制品对食用者健康影响的报道尚少。
人体肠道中栖息着大量的共生菌群,且越来越多的研究证实肠道微生物和人体的健康息息相关,与肥胖[5]及痛风[6]、炎性肠病[7]、自闭症[8]和糖尿病[9]等疾病的发生存在一定关系。同时肠道菌群亦易受膳食[10]、抗生素[11]和益生菌制剂[12]的影响。
近年来关于食用调味面制品是否对人体健康会产生影响存在较大的争议,但多数观点缺乏实验验证和数据支撑。众所周知,调味面制品在被食用3~4 h后便进入肠道进行消化,同时其消化物可能对肠道微生物产生一定的影响,然而目前关于调味面制品对食用者肠道菌群影响的研究尚未见报道。本研究采用Miseq高通量测序技术,评价了食用调味面制品对青年志愿者肠道菌群多样性的影响,以期为后续调味面制品的营养价值和安全性评价提供参考。
1 材料与方法
1.1材料与仪器
调味面制品 从襄阳市隆中鑫源超市和学子超市购买7个品名的调味面制品,每袋产品包装标注净重均为158 g;QIAamp DNA Stool Mini Kit粪便DNA基因组提取试剂盒 德国QIAGEN公司;5×TransStartTM FastPfu Buffer、FastPfu Fly DNA Polymerase、dNTPs Mix 北京全式金生物技术有限公司;DNA 1000试剂盒 美国Agilent公司。
5810R台式高速冷冻离心机 德国Eppendorf公司;UVPCDS8000凝胶成像分析系统 美国BIO-RAD公司;ND-2000C微量紫外分光光度计 美国Nano Drop公司;vetiri梯度基因扩增仪 美国AB公司;DYY-12电泳仪 北京六一仪器厂;2100芯片生物分析仪 美国Agilent公司;Miseq高通量测序平台 美国Illumina公司。
1.2实验方法
1.2.1 志愿者招募 本研究共招募志愿者9名,编号为1#~9#,其中1#~5#为女性,6#~9#为男性,在实验前每位志愿者均签署了知情同意书。志愿者年龄在19~21岁,体重正常(BMI=18.5~24.9 kg·m-2),近一个月未服用抗生素或发酵类食品。
1.2.2 调味面制品食用及样品采集 每位志愿者每隔一天食用一包调味面制品,持续2周累计食用7包,且不同品名调味面制品食用顺序一致。分别于食用前一日(食用前组),食用完7包调味面制品次日(食用2周组),及最后一次食用调味面制品后2周(停食后2周组)采集志愿者粪便样品,置于-80 ℃保藏备用。
1.2.3 DNA提取和PCR扩增 使用粪便DNA提取试剂盒提取微生物宏基因组,完整度、纯度及浓度均满足后续实验要求的DNA样品暂存到-20 ℃冰箱备用。
PCR扩增体系为:5×PCR缓冲液4 μL,2.5 mmol/L dNTPs mix 2 μL,5 μmol/L正向引物0.8 μL,5 μmol/L反向引物0.8 μL,5 U/μL DNA聚合酶0.4 μL,DNA模板10 ng,体系用ddH2O补充至20 μL。其中正向引物为338F(5′-ACTCCTACGGG AGGCAGCA-3′),反向引物为806R(5′-GGACTACH VGGGTWTCTAAT-3′),且在正向引物中加入7个核苷酸标签(barcode)。
扩增条件为:首先95 ℃变性3 min,然后95 ℃变性30 s,55 ℃延伸30 s,72 ℃退火45 s后重复35个循环,最后72 ℃延伸10 min,PCR产物置-20 ℃冰箱备用。
1.2.4 样品平衡及Miseq高通量测序 使用Agilent DNA 1000试剂盒,按照100 nmol/L的浓度对DNA模板进行稀释,寄往上海美吉生物医药科技有限公司使用Miseq平台进行高通量测序。
1.2.5 序列优化及质控 Miseq测序得到的是双端序列数据,首先根据成对序列之间的重叠(overlap)关系,将其拼接(merge)成一条序列,继而从序列尾部设置50 bp的窗口并计算该窗口内碱基质量值的平均值,若该值小于20则切掉窗口内最后一个碱基,同时窗口向序列头部移动一个碱基,并继续计算窗口内50 bp碱基的平均值,直至窗口内碱基平均得分大于20为止,然后根据barcode将所有序列划分到各个样品,并对序列方向进行校正,最后使用切掉barcode和引物的序列进行生物信息学分析。在序列拼接过程中,若overlap区的碱基数小于10 bp或最大错配比率大于0.2,则该对序列予以剔除。拼接好的序列若存在barcode碱基错配或引物碱基错配数大于2 bp,则亦予以剔除。同时本研究要求切掉barcode和引物后的序列其碱基数大于等于50 bp。
1.2.6 生物信息学分析 质控后的序列使用QIIME(v1.7.0)分析平台进行物种和多样性分析[13],主要的处理流程为:
使用PyNAST校准排齐序列[14],在序列100%相似性下进行UCLUST归并,建立无重复的16s rRNA全长序列集[15];
在序列97%相似度下进行分类操作单元(Operational taxonomic units,OTU)划分;
使用ChimeraSlayer去除可能存在嵌合体的OTU[16];
使用RDP(Ribosomal Database Project,Release 11.5)[17]和Greengenes(Release 13.8)[18]数据库对OTU代表性序列进行同源性比对,整合两个数据库的比对结果,确定每个OTU最终的分类学地位;
使用FastTree软件[19]绘制基于OTU代表性序列的系统发育进化树,随后计算样品的α和β多样性。
1.2.7 多元统计学分析 使用配对t检验对食用调味面制品不同阶段志愿者肠道菌群的丰富度指标、多样性指标和基于门属分类地位的细菌相对含量进行显著性分析,使用维恩(Venn)图对不同分组样品中特有或共有的OTU数量进行表示;使用基于UniFrac距离的主坐标分析(Principal Coordinate Analysis,PCoA)和非参数多元方差分析(PERMANOVA)对食用调味面制品不同阶段志愿者肠道菌群群落结构进行分析。使用Origin 8.5软件和R软件(v3.3.2)对QIIME产生的结果进行分析和可视化处理。
1.2.8 核酸登录号 本研究中所有序列数据已提交至MG-RAST数据库,登录号为mgp21723。
2 结果与分析
2.1序列丰富度和多样性分析
纳入本研究的27个样品16S rRNA测序情况及各分类地位数量如表1所示。
由表1可知,通过Miseq高通量测序,本研究采集的27个样品共产生了980088条高质量16S r RNA序列,平均每个样品产生36300条(范围为30289~44263,标准差为4250)。经过100%序列鉴定聚类分析后,本研究共得到497684条代表性序列,根据序列的97%相似性进行操作分类单元划分后,共得到87973个OTU,经嵌合体检测后去除了45.29%的OTU,还剩余48128个OTU,每个样品平均剩余3700个OTU(范围为2225~7262,标准差为1326)。
本研究将所有的序列鉴定为28个门,76个纲,119个目,236个科和498个属。值得一提的是,本研究有45.97%的OTU和16.44%的序列(范围为6.53%~47.30%,标准差为10.48%)不能鉴定到属水平。
只有尽可能多的获取高质量的16s rRNA序列,才可以更全面的捕获样本中的微生物信息,因而本研究产生的序列数是否满足后续生物信息学的分析,是一个值得关心的问题[20]。研究人员通常采用稀疏曲线和香农指数图对样本中微生物的丰富度和多样性进行了评价,纳入本研究27 个样品的稀疏曲线和香农指数图如图1所示。
由图1(A)可知,虽然随着序列数的增多本研究捕获新物种的数量亦随之增加,然而由图1(B)可知,每个样品的香农多样性曲线已经进入平台期。由此可见,随着测序量的进一步增加尽管新的微生物种系型可能会被发现,但微生物的多样性已经不再随之发生变化了,因而本研究的测序量是满足后续生物信息学分析要求的[21]。
由图1亦可知,每2 d食用150 g左右调味面制品持续2周及停食2周后志愿者肠道菌群的丰富度和多样性均高于食用前,这在一定程度上说明食用调味面制品可能增加了志愿者肠道菌群的丰富度和多样性。Chao1和发现物种数(Observed speices)指数常用来计算样本中菌群的丰富度,而香农(Shannon)和辛普森(Simpson)指数常用来计算多样性[22]。由表1亦可知,单个样品用于划分OTU的最小序列数为19706条,本研究进一步计算了在测序量达到该数值时,各样品中微生物的Chao1、Observed speices、Shannon和Simpson指数,其结果如表2所示。
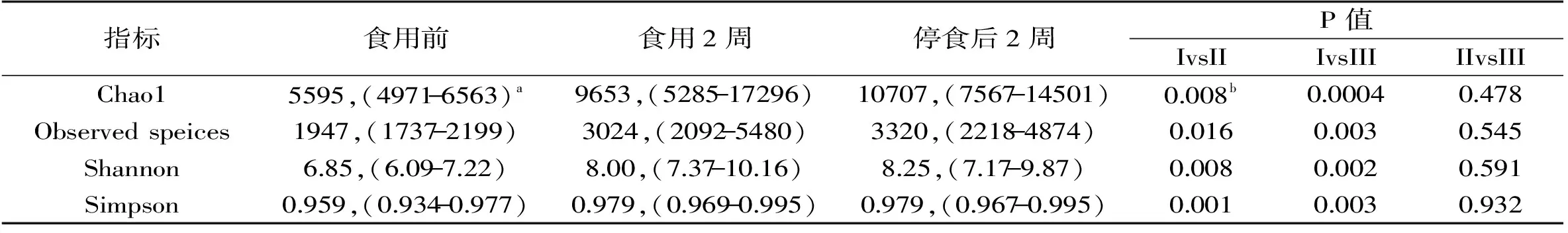
表2 食用调味面制品不同阶段志愿者肠道菌群丰富度和多样性指数比较分析Table 2 Comparison of richness and diversity indices among gut microbiota of volunteers in different stages of consumption of seasoned flour products

图1 稀疏曲线图(A)和香农指数图(B)Fig.1 Rarefaction analysis(A)and shannon diversity estimates(B)of the high throughput sequencing
注:a,中位数,(最小值-最大值);b,表中I、II和III分别代表食用调味面制品前、食用2周和停食后2周。
由表2可知,每2 d食用150 g左右调味面制品持续2周后志愿者肠道菌群的丰富度和多样性均显著增大(p<0.05),且停食2周后肠道菌群的丰富度和多样性较之食用期间均不会发生显著的变化(p>0.05)。
2.2基于OTU水平食用调味面制品对肠道菌群影响的分析
由序列丰富度和多样性分析可知,每2 d食用150 g左右调味面制品持续2周后志愿者肠道菌群的丰富度和多样性会随之增大,因α多样性分析是基于OTU水平的,故而本研究进一步在OTU水平上探讨了食用调味面制品对肠道菌群的影响。基于OTU水平的Venn图如图2所示。
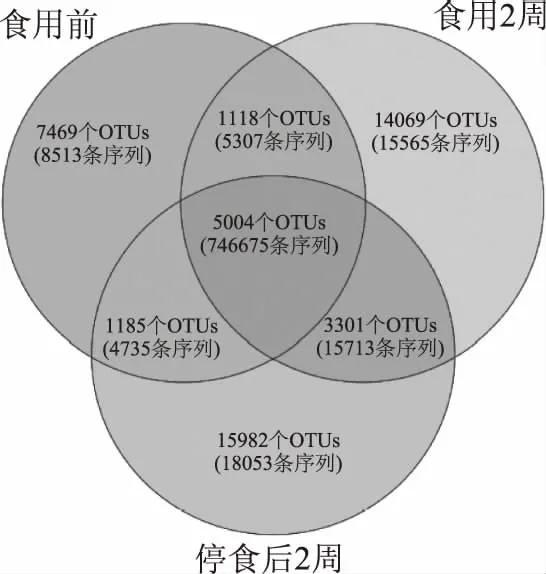
图2 基于OTU水平的Venn图Fig.2 Venn diagram based on OTU table
由图2可知,在食用调味面制品不同阶段样品中均至少出现一次的OTU共有5004个,仅占OTU总数的10.40%,而这些OTU所包含的序列数为746675条序列,占所有质控后合格序列数的76.18%。其中,27个样品均含有44个OTU,仅占OTU总数的0.091%,但其包含的序列数为163324 条序列,占所有质控后合格序列数的16.66%。值得一提的是,仅在调味面制品食用前、食用2周或停食2周样品中出现的OTU数分别为7469个、14069个和15982个,各占OTU总数的15.52%、29.23%和33.21%,其所包含序列数分别为8513条、15565条和18053条,仅占所有质控后合格序列数的0.87%、1.59%和1.84%。本研究进一步统计了OTU在27个样品中出现的次数,其结果如图3所示。
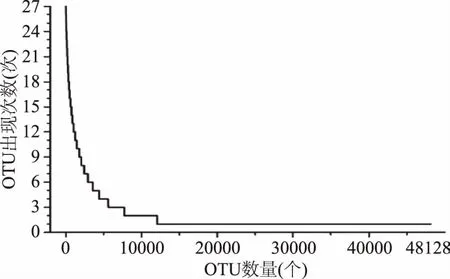
图3 OTU在27个样品中出现次数统计Fig.3 Distribution of OTU as a function of their prevalence in the 27 individuals
由图3可知,纳入本研究的27个样品共产生48128个OTU,然而在27个样品中仅出现1 次的OTU为36066个,占OTU总数的74.93%,所包含序列数为38270条,仅占所有质控后合格序列数的3.90%。值得一提的是,在27个样品中仅出现1次的OTU平均每个含有1.1条序列,以本研究中每个样品平均产生36300条序列计算,每个仅出现1次的OTU其平均相对含量仅为0.0030%。由此可见,虽然食用调味面制品后肠道菌群的种系型增多了,但所增加的种系型其相对含量是极低的。
2.3基于门属水平食用调味面制品对肠道菌群影响的分析
纳入本研究的27位志愿者肠道菌群含量最多的4个细菌门分别为Firmicutes(55.52%)、Bacteroidetes(30.00%)、Proteobacteria(5.82%)和Actinobacteria(4.08%),其余24个细菌门的相对含量累计为2.57%。经配对t检验发现,使用调味面制品不同阶段,上述28个细菌门的相对含量差异均不显著(p>0.05)。使用高通量测序技术对样本微生物多样性进行研究时,通常将相对含量大于1.0%的属定义为优势菌属,且不同群体间菌属相对含量的差异性分析亦主要基于优势菌属进行[23-24]。基于属水平食用调味面制品不同阶段志愿者肠道中细菌相对含量的比较分析如图4所示。
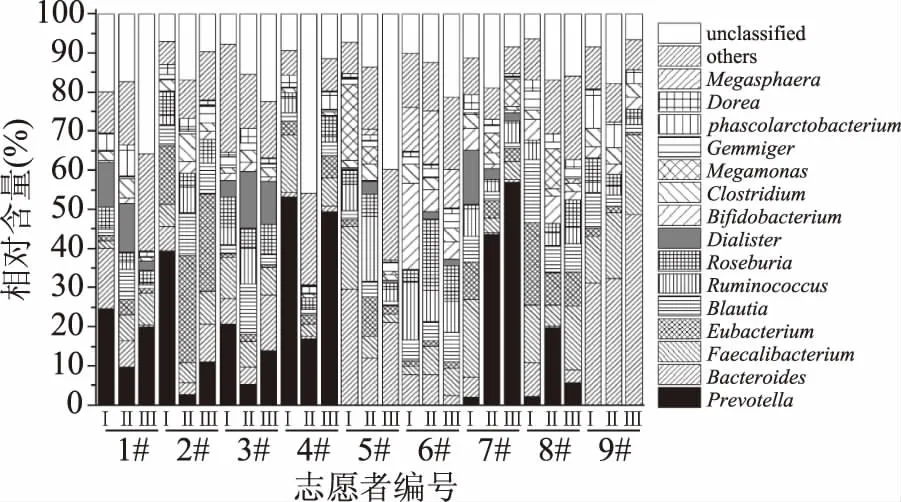
图4 基于属水平食用调味面制品不同阶段志愿者 肠道中细菌相对含量的比较分析Fig.4 Relative abundances of the major bacterial genera among gut microbiota of volunteers in different stages of consumption of seasoned flour products
由图4可知,纳入本研究的青年志愿者肠道中隶属于Firmicutes且相对含量大于1.0%的细菌属及其含量分别为:Faecalibacterium(8.89%)、Eubacterium(6.14%)、Blautia(5.15%)、Ruminococcus(4.58%)、Roseburia(4.40%)、Dialister(3.09%)、Clostridium(2.77%)、Megamonas(1.80%)、Gemmiger(1.48%)、Phascolarctobacterium(1.47%)、Dorea(1.42%)和Megasphaera(1.34%)。隶属于其他细菌门且相对含量大于1.0%的还包括:Actinobaceria的Bifidobacterium(2.82%);Bacteroidetes的Prevotella(14.68%)和Bacteroides(10.41%)。综上所述,上述15个优势细菌属中14个隶属于Firmicutes或者Bacteroidetes,其相对含量累计为67.61%。由此可见,纳入本研究的青年志愿者肠道菌群主要是由若干个隶属于硬壁菌门和拟杆菌门已知的优势细菌属组成,这与以往关于中国健康人群肠道菌群构成的研究报道结论相一致[25]。经配对t检验发现,使用调味面制品不同阶段,上述15个优势细菌属的相对含量差异均不显著(p>0.05)。由此可见,食用150 g左右调味面制品持续2周不会对青年志愿者肠道中优势菌属的相对含量产生影响。
通过配对t检验,本研究发现食用150 g左右调味面制品持续2周后青年志愿者肠道中隶属于Actinomyces、Alkaliphilus、Allochromatium、Bacillus、Cronobacter、Desulfomicrobium、Fusibacter、Ignavibacterium、Leptothrix、Paenibacillus和Reyranella的细菌其相对含量会发生显著的变化(p<0.05)。值得一提的是,上述11个细菌属在志愿者肠道中的平均含量均小于1%,且范围在0.013%~0.204%之间,不属于优势细菌属,因而本研究不再对其进行进一步的探讨。
2.4食用调味面制品对青年志愿者肠道菌群群落结构的影响
本研究进一步采用两步UCLUST法构建了OTU矩阵,并采用基于UniFrac距离的PCoA对食用调味面制品不同阶段志愿者肠道菌群群落结构变化进行了分析,其结果如图5所示。

图5 基于UniFrac距离的非加权(A)和加权(B) PCoA的志愿者肠道菌群群落结构分析Fig.5 PCoA of gut microbiota of volunteers based on unweighted and weighted UniFrac
由图5可知,基于UniFrac距离的非加权和加权的主坐标分析其第一主成分和第二主成分的贡献率分别为12.90%和5.65%、35.98%和24.75%,不同志愿者食用调味面制品同一阶段的样品均未呈现出明显的聚类趋势。值得一提的是,同一志愿者食用调味面制品不同阶段的样品呈现出了一定的聚类趋势,其中较为明显的为9#志愿者。因而我们可以定性的认为每2 d食用150 g左右调味面制品持续2周可能对志愿者的肠道菌群群落结构影响较小,其影响远小于志愿者个体之间的差异。为了对这一结论进行验证,本研究采用基于Meta-Storm距离的PERMANOVA对调味面制品食用情况、个体差异及性别对青年志愿者肠道菌群变化的影响进行了定量分析,其结果如图6所示。
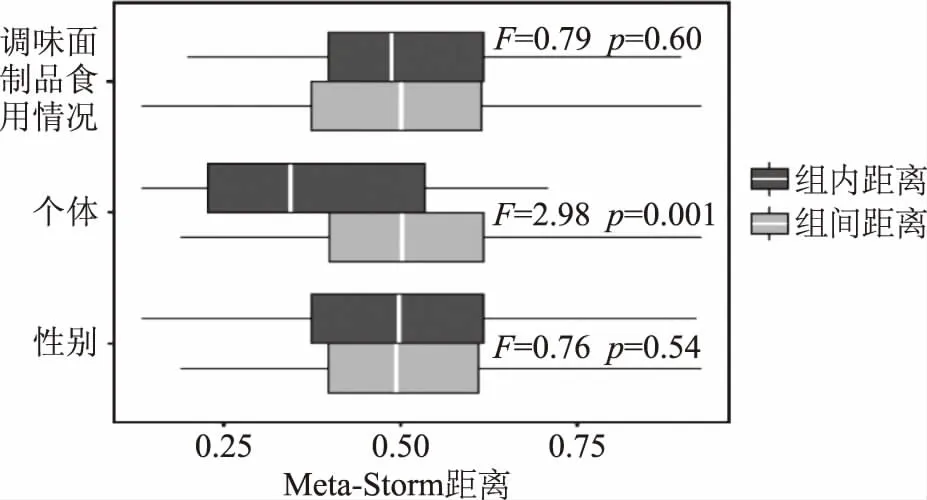
图6 不同因素对青年志愿者肠道菌群变化的影响Fig.6 Contributions of different factors on young volunteers gut microbiota variation
由图6可知,调味面制品食用情况对志愿者肠道菌群变化影响不显著(F=0.79,p=0.60)。由此可见,每2 d食用150 g左右调味面制品持续2周不会对青年志愿者肠道菌群的多样性产生影响。由图6亦可知,性别因素不会对志愿者的肠道菌群群落结构变化产生显著影响(F=0.76,p=0.54),而志愿者个体之间肠道菌群群落结构差异极显著(F=2.98,p<0.001),这均与基于UniFrac距离的PCoA结果一致。
3 讨论
由序列丰富度和多样性分析发现,食用调味面制品可以显著提高志愿者肠道菌群的丰富度和多样性,而经PCoA和PERMANOVA均发现每2 d食用150 g左右调味面制品持续2周对志愿者的肠道菌群群落结构没有显著的影响。本研究认为造成此种现象的主要原因在于,食用调味面制品后志愿者肠道菌群的种系型增加了,但由于种系型的相对含量较低因而无法对肠道菌群群落结构的整体变化产生影响。随着食用调味面制品时间的延长或食用量的增大,这些种系型的含量可能会随之增加,亦可能最终会改变食用者肠道菌群的群落结构,但该推论尚需进一步的实验验证。此外,由于调味面制品生产工艺简单、设备投入低,因而调味面制品生产企业存在门槛低、产品质量参差不齐的特点,然而由于各种因素限制,本研究仅采集了7个品名的调味面制品,且生产企业在行业内均具有较高的知名度,这也可能是导致上述现象发生的原因之一。
4 结论
每2 d食用150 g左右调味面制品持续2周后,青年志愿者肠道菌群的丰富度和多样性均显著增大,但由于增加种系型的相对含量较低,而青年志愿者肠道中优势菌属的相对含量和菌群结构均未发生显著变化。
[1]刘超,罗红霞,句荣辉,等. 分光光度法间接测定调味面制品中甜蜜素的含量[J]. 中国食品添加剂,2013,20(1):222-226.
[2]安虹,谭炜. 调味面制品产品质量现状调研及分析[J]. 安徽农业科学,2016,44(3):88-89.
[3]刘畅. 高盐高油无国标辣条年售500亿元[J]. 中国质量万里行,2016,23(2):26-29.
[4]刘少伟,阮赞林. 疯狂的“辣条”——有关“辣条”的食品安全隐患[J]. 质量与标准化,2015,22(5):35-36.
[5]Daniel H,Gholami A M,Berry D,et al. High-fat diet alters gut microbiota physiology in mice[J]. The ISME journal,2014,8(2):295-308.
[6]Guo Z,Zhang J,Wang Z,et al. Intestinal microbiota distinguish gout patients from healthy humans[J]. Scientific reports,2016,6(2):1-10.
[7]Wang T,Cai G,Qiu Y,et al. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers[J]. The ISME journal,2012,6(2):320-329.
[8]De Theije C G M,Wopereis H,Ramadan M,et al. Altered gut microbiota and activity in a murine model of autism spectrum disorders[J]. Brain,behavior,and immunity,2014,37(3):197-206.
[9]Forslund K,Hildebrand F,Nielsen T,et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota[J]. Nature,2015,528(7581):262-266.
[11]Russell S L,Gold M J,Reynolds L A,et al. Perinatal antibiotic-induced shifts in gut microbiota have differential effects on inflammatory lung diseases[J]. Journal of Allergy and Clinical Immunology,2015,135(1):100-109.
[12]Kwok L Y,Wang L,Zhang J,et al. A pilot study on the effect of Lactobacillus casei Zhang on intestinal microbiota parameters in Chinese subjects of different age[J]. Beneficial microbes,2014,5(3):295-304.
[13]Caporaso J G,Kuczynski J,Stombaugh J,et al. QIIME allows analysis of high-throughput community sequencing data[J]. Nature methods,2010,7(5):335-336.
[14]Caporaso J G,Bittinger K,Bushman F D,et al. PyNAST:a flexible tool for aligning sequences to a template alignment[J]. Bioinformatics,2010,26(2):266-267.
[15]Edgar R C. Search and clustering orders of magnitude faster than BLAST[J]. Bioinformatics,2010,26(19):2460-2461.
[16]Haas B J,Gevers D,Earl A M,et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons[J]. Genome research,2011,21(3):494-504.
[17]Cole J R,Chai B,Farris R J,et al. The ribosomal database project(RDP-II):introducing myRDP space and quality controlled public data[J]. Nucleic acids research,2007,35(1):D169-D172.
[18]Desantis T Z,Hugenholtz P,Larsen N,et al. Greengenes,a chimera-checked 16S rRNA gene database and workbench compatible with ARB[J]. Applied and environmental microbiology,2006,72(7):5069-5072.
[19]Price M N,Dehal P S,Arkin A P. Fasttree:computing large minimum evolution trees with profiles instead of a distance matrix[J]. Molecular biology and evolution,2009,26(7):1641-1650.
[20]Caporaso J G,Lauber C L,Walters W A,et al. Ultra-high-throughput microbial community analysis on the IlluminaHiSeq and MiSeq platforms[J]. The ISME journal,2012,6(8):1621-1624.
[21]Xu J,Lian F,Zhao L,et al. Structural modulation of gut microbiota during alleviation of type 2 diabetes with a Chinese herbal formula[J]. The ISME journal,2015,9(3):552-562.
[22]Ahrén I L,Xu J,Önning G,et al. Antihypertensive activity of blueberries fermented by Lactobacillus plantarum DSM 15313 and effects on the gut microbiota in healthy rats[J]. Clinical Nutrition,2015,34(4):719-726.
[23]Sommer F,Bäckhed F. The gut microbiota-masters of host development and physiology[J]. Nature Reviews Microbiology,2013,11(4):227-238.
[24]Kamada N,Seo S U,Chen G Y,et al. Role of the gut microbiota in immunity and inflammatory disease[J]. Nature Reviews Immunology,2013,13(5):321-335.
[25]Zhang J,Guo Z,Xue Z,et al. A phylo-functional core of gut microbiota in healthy young Chinese cohorts across lifestyles,geography and ethnicities[J]. The ISME journal,2015,9(9):1979-1990.
Effectofconsumptionofseasonedflourproductsongutmicrobiotainyoungvolunteers
CAIHong-yu1,WANGYan2,SHENXin1,MAXue-wei3,WANGDan-dan1,YUHai-zhong1,GUOZhuang1,*
(1.Northwest Hubei Research Institute of Traditional Fermented Food,School of Chemical Engineering and Food Science,Hu Bei University of Arts and Science,Xiangyang 441053,China; 2.Inspection Department,Dangyang City Maternal and Child Health Hospital,Yichang 444100,China; 3.Xiangyang Entry-exit Inspection and Quarantine Bureau Comprehensive Laboratory,Xiangyang 441003,China)
In the current study,9 young volunteers were recruited and each subject consumption of a packet of seasoned flour products with the weight of 150 g for a continuous period of 2 weeks. Meanwhile,gut microbiota of subjects were monitored before starting,during and after stopping consumption for 2 weeks using high throughput sequencing technologies. The result indicated that consumption of seasoned flour products for 2 weeks significantly increased the gut microbiota diversity and richness by paired t test(p<0.05),but had no influence on the relative abundance of domain bacterial genus(p>0.05). Through principal coordinate analysis based on UniFrac and PERMANOVA based on Meta-Storm,it was shown that the consumption of seasoned flour products for 2 weeks couldn’t alter the structural of gut microbiota.
seasoned flour products;high throughput sequencing;gut microbiota;young
2017-04-18
蔡宏宇(1996-),女,本科生,研究方向:食品生物技术,E-mail:1486721051@qq.com。
*
郭壮(1984-),男,博士,副教授,研究方向:食品生物技术,E-mail:guozhuang1984@163.com。
湖北省荆楚卓越工程师协同育人计划(201657);湖北文理学院大学生创新创业训练计划项目(2017)。
TS201.4
A
1002-0306(2017)21-0289-07
10.13386/j.issn1002-0306.2017.21.057
猜你喜欢
中老年保健(2022年2期)2022-08-24
江苏调味副食品(2022年1期)2022-05-17
中国饲料(2022年5期)2022-04-26
中国食品(2021年15期)2021-08-27
食品安全导刊(2020年21期)2020-12-03
科学(2020年4期)2020-11-26
江苏调味副食品(2020年2期)2020-06-23
益寿宝典(2018年2期)2018-08-20
美食(2017年5期)2017-05-11
现代面粉工业(2016年2期)2016-02-23
