
三氟甲磺酸三甲基硅酯的合成工艺研究
2017-11-13 00:51王少波杨献奎吕灵华
低温与特气 2017年5期
李 林,王少波,杨献奎,吕灵华
(中国船舶重工集团公司 第七一八研究所,河北 邯郸 056107)
·工艺与设备·
三氟甲磺酸三甲基硅酯的合成工艺研究
李 林,王少波,杨献奎,吕灵华
(中国船舶重工集团公司 第七一八研究所,河北 邯郸 056107)
以三氟甲磺酸和三甲基氯硅烷为原料,合成三氟甲磺酸三甲基硅酯。通过考察温度、时间、原料比例、蒸馏压力等影响因素,确定了最佳的反应条件:温度20~30℃,反应时间8 h,原料摩尔比1:1.40,减压蒸馏压力-0.090MPa。实验利用了核磁共振谱图(1H,19F,29Si NMR)和化学滴定法对目标产物进行了定量分析,产品收率可以达到98%以上,纯度可达99%以上。
三氟甲磺酸三甲基硅酯;三氟甲磺酸;三甲基氯硅烷;合成
0 引 言
三氟甲磺酸硅烷基衍生物广泛应用于化工医药行业,主要包括三氟甲磺酸三甲基硅酯、三氟甲磺酸三异丙基硅酯、三氟甲磺酸三乙基硅酯、三氟甲磺酸叔丁基二甲硅酯等,其中以三氟甲磺酸三甲基硅酯应用最为广泛[1]。
三氟甲磺酸三甲基硅酯,分子式CF3SO3Si(CH3)3,英文名Trimethylsilyl Trifluormethane -sulfonate, 缩写为TMSOTf,外观无色透明液体,在空气中极易水解发烟,有刺激性气味。三氟甲磺酸三甲基硅酯是一种十分有效的甲基硅烷基化试剂,常用于作为羟基、羧基、羰基等官能团转化与保护[2-3],并参与碳碳成键增长碳链的反应,又可用于Lewis酸催化剂[4]及作为阳离子引发剂[5-6],是重要的医药中间体[7-8]和有机合成结构单元。
德国科学家在上世纪70年代首次公开报道用三氟甲磺酸银和三甲基氯硅烷反应制备三氟甲磺酸三甲基硅酯[9],80年代又有报道用三氟甲磺酸酐和三甲基硅醚制备产品[10]等方法,这些方法原料成本较高,不适合工业化生产,之后又发展了三氟甲磺酸和四甲基硅烷反应的工业化制备,但原料四甲基硅烷在制备过程往往带有大量的二甲基丁烷、二甲基氯硅烷、甲基二硅烷等杂质,这些杂质会与三氟甲磺酸反应生成副产物。尤其是甲基二氯硅烷和三氟甲磺酸也会生成三氟甲磺酸甲基硅酯与三氟甲磺酸三甲基硅酯沸点接近,普通精馏方法很难分离,给三氟甲磺酸三甲基硅酯的生产纯化带来很大困难。陈红斌等[11]公开了一种三氟甲磺酸盐与硅烷化试剂反应,可以获得高纯度产品,但由于原料为酸盐,硅烷化试剂过量导致生产成本较高。
本文以三氟甲磺酸和三甲基氯硅烷为原料,深入对反应的各个影响条件进行试验优化,探索高收率合成三氟甲磺酸三甲基硅酯的工艺。
1 试验部分
1.1反应方程式

1.2试剂与仪器
无水三氟甲磺酸(CF3SO3H)、无水三甲基氯硅烷((CH3)3SiCl):分析纯,梯希爱(上海)化成工业发展有限公司;
NaOH:分析纯,国药集团化学试剂有限公司;
德国Bruker公司AV300MHz核磁共振波谱仪,CDCl3为氘代溶剂,TMS为内标;
上海菁华科技仪器有限公司7230G可见分光光度计。
1.3实验步骤
在500 mL三口烧瓶中装入150.0 g(1.0 mol)三氟甲磺酸,用恒压滴液漏斗将150.0 g(1.38 mol,1.38 eq)三甲基氯硅烷缓缓滴入三口瓶中,室温搅拌,用碱液瓶进行尾气吸收处理,反应开始碱液瓶有大量气泡冒出,至三甲基氯硅烷滴加结束,继续搅拌反应2~10 h,减压蒸馏,收集中馏分,即得到目标产物三氟甲磺酸三甲基硅酯。
2 分析与表征
采用核磁共振谱图定性分析产品,核磁结果为1H NMR (300 MHz, CDCl3) δ 0.49 (s, 9H),19F NMR (282 MHz, CDCl3) δ -77.62 (s, 3F),29Si NMR (99 MHz, CDCl3) δ 43.98 (s, 1Si)。查询相关资料[12],并与标准样谱图进行比对,可确定为产品样。
采用化学滴定法定量分析产品。在锥形瓶中,加入30 mL水,称量质量为m的产品,静置30 min,待完全溶解后,加入两滴0.1%的酚酞试剂,用标定好的浓度为c的NaOH标准溶液进行滴定,消耗标准溶液体积为V,利用给定酸碱滴定公式计算产品的纯度。
氯含量的测定。氯含量高会影响产品质量,由原料可知产品中氯应来自原料三甲基氯硅烷中,由于三甲基氯硅烷极易水解并解离出氯离子[13],所以可用可见分光光度计检测氯化银沉淀法来检测产品中氯含量。

表1 原料及产物物性表
3 结果与讨论
3.1反应温度的影响
固定三氟甲磺酸质量为150.0 g(1.0 mol),三甲基氯硅烷质量为150.0 g(1.38 mol,1.38 eq),反应平衡时间为8 h,考察反应温度对产品收率的影响,实验结果见图1。由图1可得,首先,反应收率随着温度的升高而升高,其次,当温度达到15℃时,反应收率达到平稳,最后继续升高温度至45℃,反应收率随着温度的升高有所降低。可能的原因,反应为吸热反应,当温度升高,有利于反应的进行,所以反应收率随温度的升高而升高。当温度达到20~30℃,反应收率达到平衡,收率达到最佳。继续升高温度至30~45℃,由于三甲基氯硅烷沸点低,如果该反应过快会迅速生成氯化氢气体,气体逸出会载带部分三甲基氯硅烷,致使原料损失,影响产品的收率。因此,反应温度为20~30℃时最理想。

图1 反应温度对产品收率的影响
3.2反应时间的影响
固定三氟甲磺酸质量为150.1 g(1.0 mol),三甲基氯硅烷质量为150.0 g(1.38 mol,1.38 eq),反应温度为20℃,考察反应时间对产品收率的影响,结果见图2。由图2可得,随着反应时间的延长,三氟甲磺酸三甲基硅酯产品收率逐渐增加;反应时间在1~8 h时,产品收率增加较显著,8 h之后,产品收率维持稳定。说明反应时间为8 h之后,反应已接近进行完全。即反应原料滴加结束还需继续反应8 h才能反应彻底,可能原因是因为滴加过程中体系内会产生大量的氯化氢,虽然大量逸出,但仍有少量氯化氢气体残留在体系内,抑制反应的进行,需要延长反应时间提高反应转化率,实验确定最佳反应时间为8 h。

图2 反应时间对产品收率的影响
3.3反应比例的影响
固定三氟甲磺酸质量为150.0 g(1.0 mol),反应温度为20℃,三甲基氯硅烷的量分别按如表中摩尔比加入(以三氟甲磺酸为1),考察反应加料摩尔比对产品收率的影响,结果见图3。由图3左侧可见,三氟甲磺酸与三甲基氯硅烷摩尔比为1:0.90~1:1.50,随着三甲基氯硅烷用量的增加,产品硅酯的收率逐渐增加;当原料摩尔比达1:1.40时,收率可达98%,并维持稳定,可能的原因是,三甲基氯硅烷在体系内因其沸点偏低并有载带损失,所以使其过量才能保证三氟甲磺酸反应完全,提高产品收率。由图3右侧可见,原料摩尔比在1:0.90~1:1.40时,产品中氯含量也相对较低,原料摩尔比1:1.40之后氯含量呈上升趋势;可能的原因是,三甲基氯硅烷过量太多之后,会残留在体系内,在后期收集产品馏分的时夹带进入产品,造成氯含量偏高,影响产品质量,同时造成原料浪费。结合产品的质量要求氯含量在200×10-6以下,并保持较高的收率。因此,选择最佳原料(酸烷比)摩尔比为1:1.40。

图3 反应摩尔比对产品收率的影响
3.4蒸馏压力的影响
固定三氟甲磺酸为150.0 g(1.0 mol),三甲基氯硅烷为150.0 g(1.38 mol,1.38 eq),反应温度为20℃,考察减压蒸馏压力对产品收率的影响,由于减压蒸馏下压力为负压,本实验的压力以真空度表示,结果见图4。由图4可得,主要考察了减压蒸馏真空度为0.050~0.095 MPa,产品收率及对应的产品沸点的影响。在考察范围内,产品收率趋势平稳,均保持在98%以上;减压蒸馏产品沸点随着真空度的提高有明显下降趋势。由于蒸馏操作本身能耗较大,蒸馏沸点越高,能耗越大,采用高真空,低沸点出料成本较小,结合工艺特点及便捷性[14],实验确定以减压蒸馏真空度为0.09 MPa,即蒸馏压力为-0.09 MPa最为理想。
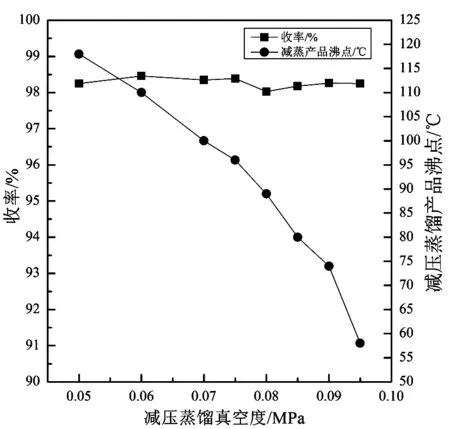
图4 减压蒸馏真空度对产品收率的影响
3.5催化剂的影响
实验分别考察了加入DMAP,三乙胺为催化剂[15]各三组实验,结果如图5所示。由图5可得,催化剂对产品收率没有明显的提高,甚至有副产物生成,考虑到工业中减压蒸馏分离过程中,增加第三组分对分离效果及其后期处理均有很大困难。本实验在保持收率稳定的情况下,暂不考虑加催化剂。

图5 催化剂对产品收率的影响
4 结论
作为医药化工中间体的三氟甲磺酸三甲基硅酯产品,以其优异的化学性能被广泛应用于合成医药等领域[16]。本研究采用三氟甲磺酸与三甲基氯硅烷反应,避免了用四甲基硅烷为原料引入的杂质,同时原料成本也相对偏低,具有市场竞争力。通过本实验,探索得到最佳反应条件为:无需催化剂,温度为20~30℃,反应时间为8 h,三氟甲磺酸与三甲基氯硅烷摩尔比为1:1.40,减压蒸馏压力为-0.090 MPa,产品收率可达98%以上,并且纯度可达99%以上。
[1] 中国船舶重工集团公司第七一八研究所.一种三氟甲磺酸三甲硅酯的制备方法:CN 103665017A[P],2014-03-26.
[2] SCHWEIZER FRANK,HINDSGAUL OLE ,et al. Synthesis of agalacto-configured C-ketoside-based γ-sugar-amino acid and its use in peptide coupling reactions[J].Carbohydrate Research,2006(341):1730-1736.
[3] PANAYIOTIS A PROCOPIOU,SIMON P D BAUGH,STEPHEN S. FLACK,et al. Inglis. An Extremely Powerful Acylation Reaction ofAlcoholswith AcidAnhydridesCatalyzed by TrimethylsilylTrifluoromethanesulfonate[J]. J Org Chem,1998(63):2342-2347.
[4] TOMOYA OGAWA, KAZUO BEPPU,et al.Trimethylsilyltrifluoromethanesulfonate asaneffective catalyst forglycoside synthesis[J]. Carbohydrate Research,1981(93):C6-C9.
[5] ROBERT F CUNICO,CHIA P KUAN,et al. Synthesis of Oxazoles from O-TrimethylsilylAcyltsime -thylsilaneCyanohydrins[J]. J.Org.Chem,1992(57):3331-3336.
[6] HERBERT M ZERTH, NICHOLAS M LEONARD, RAM S. Mohan.Synthesis of Homoallyl Ethers viaAllylationof Acetals in Ionic Liquids Catalyzed by Trimethylsilyl Trifluoromethanesulfonate [J] .Organic letters,2003,5(1):55-57.
[7] 贵州大学.地西他滨(Decitabine)的合成工艺.CN 101307084A[P],2008-11-19.
[8] TANG E, LI WEN, GAOZHANG YONG,et al.TMSOTf-catalyzed intramolecular seleno-arylation oftethered alkenes:A novel method for the solid-phasesynthesis of dihydrocoumarins and coumarins[J].Chinese Chemical Letters,2012(23):631-634.
[9] SCHMEISSER M,SARTORI P,LIPPSMEIER B. Chemische Berichte-Recueil[J].Chenische Berichet-Recuneil,,1970,103(3),868-879.
[10] JESUSM, AIZPYRUA,C PALOMO.Reagents and Synthetic Methods; 43.A New Preporntion of Trimethylsilyl Trifluromethanesulfonate[J].Synthesis-Stuttgart,1985(2):206-207.
[11] 江西国化实业有限公司.三氟甲磺酸三甲硅酯的制备方法:CN 102911196A[P],2013-02-06.
[12] 仇镇武.含氟化合物核磁共振谱图集的研究与应用[D].广州:广东工业大学.2013.
[13] 史宝萍,赵晓霞,李兴,等.三甲基氯硅烷的性质及其应用研究进展[J].化工时刊,2010,24(5):53-57.
[14] 中国船舶重工集团公司第七一八研究所.一种三氟甲磺酸三甲硅酯的纯化方法:CN 104262376A[P].2015-01-07.
[15] 李增春,边沿江,张敬周.三乙胺在三氟甲磺酸三甲硅酯反应中的作用[J].内蒙古民族师院学报(自然科学版),1997,12(2):176-180.
[16] GERT V L.Silylalling A gents[J]. Fluka Chiemie AG, 1988(1):45-47.
StudyontheSynthesisofTMSOTf
LI Lin,WANG Shaobo,YANG Xiankui,LV Linghua
(The 718th Research Institute of CSIC,Handan 056107,China)
TMSOTf was prepared,with trimethylchlorosilane and triflic acid as raw material.This work studied the influence of reaction temperature,reaction time,the ratio of acid/alkane(mol ratio) and distillation pressure. The results showed that the best conditions of the reaction temperature was 20~30℃,the time was 8h,the ratio of acid/alkane(mol ratio) was 1:1.40,and the distillation pressure was -0.09 MPa. In addition, the target product was quantitatively analyzed by1H ,19F&29Si NMR and chemical titration, which proved that the yield of the product was over 98% and the purity was over 99%.
TMSOTf;triflic acid;chlorotrimethylsilane;synthesis
2017-09-09
TQ031.2
B
1007-7804(2017)05-0034-05
10.3969/j.issn.1007-7804.2017.05.007
李林(1992),男,河南安阳人,硕士研究生,从事三氟甲磺酸及其衍生物的工业化生产工艺研究。E-mail:18833069170@163.com。
我国“乙炔制乙烯”技术获突破
2017年9月22日,由神雾科技集团研究院和北京华福工程公司联合开发的“乙炔加氢制乙烯工艺及装备”通过了中国石油和化学工业联合会组织的科技成果鉴定。业内人士表示,“乙炔加氢制乙烯技术”是现代煤化工工艺领域一项突破性技术,有望降低我国乙烯的对外依存度。
据了解,经过现场质询讨论、审阅项目研发报告和第三方检测和评价报告等环节,鉴定委员会认为神雾“乙炔加氢制乙烯工艺及装备”填补了国内外高浓度乙炔制乙烯技术空白,创新性突出。鉴定委员会由来自清华大学、石油化工科学研究院、石化规划院、天辰公司、神华研究院、中国矿业大学、山西潞安集团等单位的9名国内外知名化工专家组成,中国工程院金涌院士和舒兴田院士分别担任鉴定委员会的主任和副主任。
利用“乙炔加氢制乙烯技术及装备”建设的中试装置,经现场72小时满负荷考核表明,乙炔转化率达到98.90%,乙烯选择性达到91.06%。该成果目前申请国内外专利58项,已获授权专利32项。鉴定委员会一致认为,该技术及装备具有重要推广意义,建议加快大型工业示范装置的建设。
神雾科技集团主要从事煤炭高效清洁利用技术的开发、转化与市场推广。针对我国煤炭资源丰富特点和巨大的化工基础原料市场需求,神雾科技集团自主开发了“蓄热式电石生产新工艺”并实现了工业示范装置的连续稳定运行,拓展了乙炔下游产品深加工的路径和可能性。在此基础上,通过“乙炔加氢制乙烯技术”的深度开发,形成了独具神雾特色的乙炔法煤化工的技术体系。与传统的煤气化法煤化工相比,该技术体系具有投资强度小、能源转化效率高、水耗低、CO2排放量低、产品生产成本低等优点,实现了我国煤炭分质分级利用技术的新突破,目前多个工业示范项目正在建设中。
专家分析认为,该技术大面积推广后,能有效降低我国乙烯的对外依存度,继而减轻石脑油裂解制烯烃的产业压力,对保障我国经济健康发展、保障国家能源安全具有战略意义。鉴定委员会主任、中国工程院院士金涌表示:“该技术成果使得规模化乙炔加氢制乙烯成为可能,为我国乙烯及其下游工业的健康、可持续发展带来了新机遇,对促进我国煤化工的发展及开创乙烯原料多元化的生产路线具有重要意义。”
二氧化碳开采页岩气一举多得
近日召开的国家重点基础研发计划(973计划)“超临界二氧化碳强化页岩气高效开发基础”阶段成果总结暨研讨会上获悉,我国科学家利用超临界二氧化碳高效开发陆相页岩气,把二氧化碳压入地下封存,同时把页岩气采上来,一举多得。
此项目于2013年10月由国家科技部正式立项,武汉大学、重庆大学、陕西延长石油(集团)有限责任公司等9家单位共同承担。
据项目首席科学家李晓红院士介绍,项目研究人员通过超临界二氧化碳破岩、压裂增渗、置换页岩气机理等方面的基础研究与关键技术攻关,形成超临界二氧化碳强化页岩气高效开发理论体系和技术方法。具体技术是将液态二氧化碳注入页岩气井下。当温度和压力处于31.1℃、7.38兆帕以上时,二氧化碳就处于超临界态,此时它既有气体的低黏度、超强的流动性和渗透性,又有液体的高密度。页岩对二氧化碳的吸附能力是吸附页岩气的4—20倍,超临界二氧化碳能将井下的页岩气挤出。
今年6月,项目组在陕西延长石油—延2011井进行了我国首次页岩气超临界二氧化碳压裂现场试验,并取得圆满成功。这标志着我国在自主探索陆相页岩气高效开发方面取得了重要的理论和技术突破,在这一领域走到了国际前沿,有望开辟一条绿色、环保、高效的陆相页岩气开发新途径。
猜你喜欢
中国典型病例大全(2022年9期)2022-04-19
能源化工(2021年2期)2021-12-30
中国民间疗法(2021年17期)2021-11-04
有机氟工业(2020年2期)2020-07-04
广东石油化工学院学报(2016年6期)2016-05-17
合成化学(2015年10期)2016-01-17
合成化学(2015年5期)2015-03-26
中国当代医药(2015年9期)2015-03-01
中华皮肤科杂志(2014年4期)2014-12-19
有机氟工业(2014年3期)2014-06-05
