
杆状体肌病临床、病理及分子生物学研究进展
2017-11-03 12:39赵燕刘卓常蕾蕾卢正娟吴家勇牛丰南徐运
临床神经病学杂志 2017年5期
赵燕,刘卓,常蕾蕾,卢正娟,吴家勇,牛丰南,徐运
·综述·
杆状体肌病临床、病理及分子生物学研究进展
赵燕,刘卓,常蕾蕾,卢正娟,吴家勇,牛丰南,徐运
先天性肌病(CMs)是一组单基因遗传性骨骼肌疾病,具有临床/遗传异质性,活检骨骼肌病理存在特定的结构改变。遗传方式有常染色体显性遗传(AD)、常染色体隐性遗传(AR)、X连锁隐性遗传(XR)及散发。发病率暂无统一统计,瑞典估计约为1/22 480,北爱尔兰为1/13 500[1]。典型临床表现多数以新生儿/幼儿期发病的四肢/轴向肌群肌无力及肌张力低下,病情相对稳定/进展缓慢。可伴有面肌受累(细长脸、高颚弓等)和骨关节受累(先天性髋关节发育不良、脊柱侧弯、跟腱挛缩等)。血清肌酸激酶(CK)正常/轻度升高,EMG正常/肌源性改变。骨骼肌活检共同的病理特点:无明显肌纤维变性、坏死,可存在肌纤维类型分布异常。根据其特征性病理特点,North等[2]将CMs主要分为以下5类:(1)轴空性肌病,包括中央轴空病(CCD)和多微小轴空病(MmD);(2)中心核肌病(CNMs);(3)杆状体肌病(NM),根据杆状体不同的病理表现形式还包括帽状肌病、斑马体肌病、轴空/杆状体肌病;(4)肌球蛋白储积病(MSM)又称为透明体肌病;(5)先天性肌纤维类型不均(CFTD)。
NM是CMs中最常见的类型之一,呈AD/AR遗传。NM具有广泛的临床表现,肌无力程度轻重不等,最严重的患儿在出生后无法自主呼吸;而症状较轻的患者仅有轻度肢体无力,病情稳定/不进展[3]。
1 临床特点
根据ENMC国际协作组[4]的分型,NM主要分为以下6型:(1)先天性NM严重型(16.1%)[5],出生时即有严重的肌无力、肌张力低下,无自发运动及自主呼吸,很快死亡;(2)中间型(20.3%),较先天性NM严重型稍轻,肢体肌力可达Ⅰ级,通过呼吸支持可以维持生命,但不能行走;(3)轻症型(即经典型)(46.1%),婴儿期/儿童期起病,主要以面肌、躯干肌无力,伴肌张力低下,运动发育迟缓,病情相对稳定;(4)儿童起病严重型(13.3%);(5)散发的成人起病型(4.2%);(6)少数骨骼肌病理发现杆状体,但需诊断为骨骼肌细肌丝蛋白病或肌病伴杆状体,以区别先天性杆状体肌病。根据患者症状的严重程度对NM进行分类,有助于判断该病的预后及可能的致病基因。但随着分子生物学的发展,特定基因变异对应特定的临床、病理表型,因此目前NM的分类倾向于依据变异基因进行分类。尽管如此,某些变异基因可以有相似或共同的临床/肌群受累表现,依据临床表现,可指导基因测序方向,如ACTA-1、NEB、KLHLF40突变的NM可出现极严重的新生儿肌张力低下,表现为松软儿的特征,包括胎儿运动不能或运动功能减退、关节挛缩/骨折、呼吸衰竭,并在出生时吞咽/喂养困难[6];NEB、TPM3、TPM2突变的NM,临床表型相似,轻中度受累的患者表现出明显的颈部肌群无力和趾屈无力,可出现足下垂和弓形足。NEB-NM在轻症患者中可仅有小腿胫骨前肌受累,重症患者下肢肌肉弥漫性受累[7],头颅MRI显示选择性侵犯翼外肌,并影响咀嚼肌和舌肌[8]。TPM2-NM主要影响咀嚼肌(颞)及下肢远端比目鱼肌与远端伸、屈肌,而大腿肌群无明显受累[9]。
2 骨骼肌病理活检
该组肌病无论是哪种致病基因所致,其特征性表现均为骨骼肌活检于肌细胞胞浆中可见高密度的杆状体(MGT染色观察最为典型——红染杆状体)。杆状结构的数量可随年龄的增长而增多,但其与疾病的严重程度无关。不同部位的肌肉组织及不同肌纤维中杆状体数目不尽相同,故有些NM患者没有在杆状体聚集的部位取材可能漏诊,需行二次活检才能明确诊断[10]。杆状体通常出现在胞浆中且多位于肌膜下,极少病例(如ACTA1-NM)可于细胞核中观察到杆状体。免疫组化示杆状体与Z线具有相同的蛋白质,α-辅肌动蛋白、肌动蛋白、肌钙蛋白T、肌原肌球蛋白和结蛋白染色阳性[11-13]。电镜下杆状体呈杆状或卵圆状,与肌纤维长轴平行,由于杆状体与Z线有相同的晶格结构和蛋白质,推测某些杆状体可能来源于Z线[14]。帽状肌病曾作为一类独立肌病,后来研究[15-17]发现,帽状体表现为肌膜下边界清晰的帽状物,出现于4%到几乎100%肌纤维中,MGT染色为紫色偏蓝或绿色,HE染色为嗜酸性,NADH-TR染色为暗蓝色,ATPase染色减低,电镜下与杆状体结构一致,免疫组化与NM一致,故目前帽状肌病已归类于NM。与其他类型NM不同的是帽状体累及肌纤维的多少与疾病的严重程度和患者年龄相关。轴空结构[18-19]可以与杆状体同时存在,杆状体聚集区域缺乏线粒体,因此该区域氧化酶染色示缺如呈轴空结构。这点也与轴空性肌病的轴空形成相鉴别,轴空性肌病的轴空结构是由于肌原纤维断裂、排列紊乱而缺少线粒体。杆状体并非NM特有,亦可出现正常眼肌、老年人肌肉[20]及少数遗传性(线粒体脑肌病、强直性肌营养不良、Hodgkin病等)或获得性神经肌肉病(皮肌炎、HIV感染等[21])。
与其他CMs一样,NM可见肌纤维直径大小不等,Ⅰ型肌纤维萎缩/发育不良,在肌纤维类型上呈Ⅰ型肌纤维优势/单一Ⅰ型肌纤维。无肌纤维变性、坏死及再生。
3 分子生物学研究及发病机制
目前已发现10种基因变异(表1)可引起NM,其临床表现具有相似或者相同的症状。TPM3、NEB、ACTA1、TPM2、TNNT1、CFL2编码肌节细肌丝蛋白或相关蛋白,KBTBD13、KLHL40 (KBTBD5)和KLHL41 (KBTBD10)涉及到泛素-蛋白酶体通路(UPP),编码小于5/6结构域的KLHL蛋白,该蛋白属于标准KLHL蛋白家族(包括BTB、BACK、5/6 Kelch结构域)中的一种,又被归类于KBTBD家族[19,22-29]。
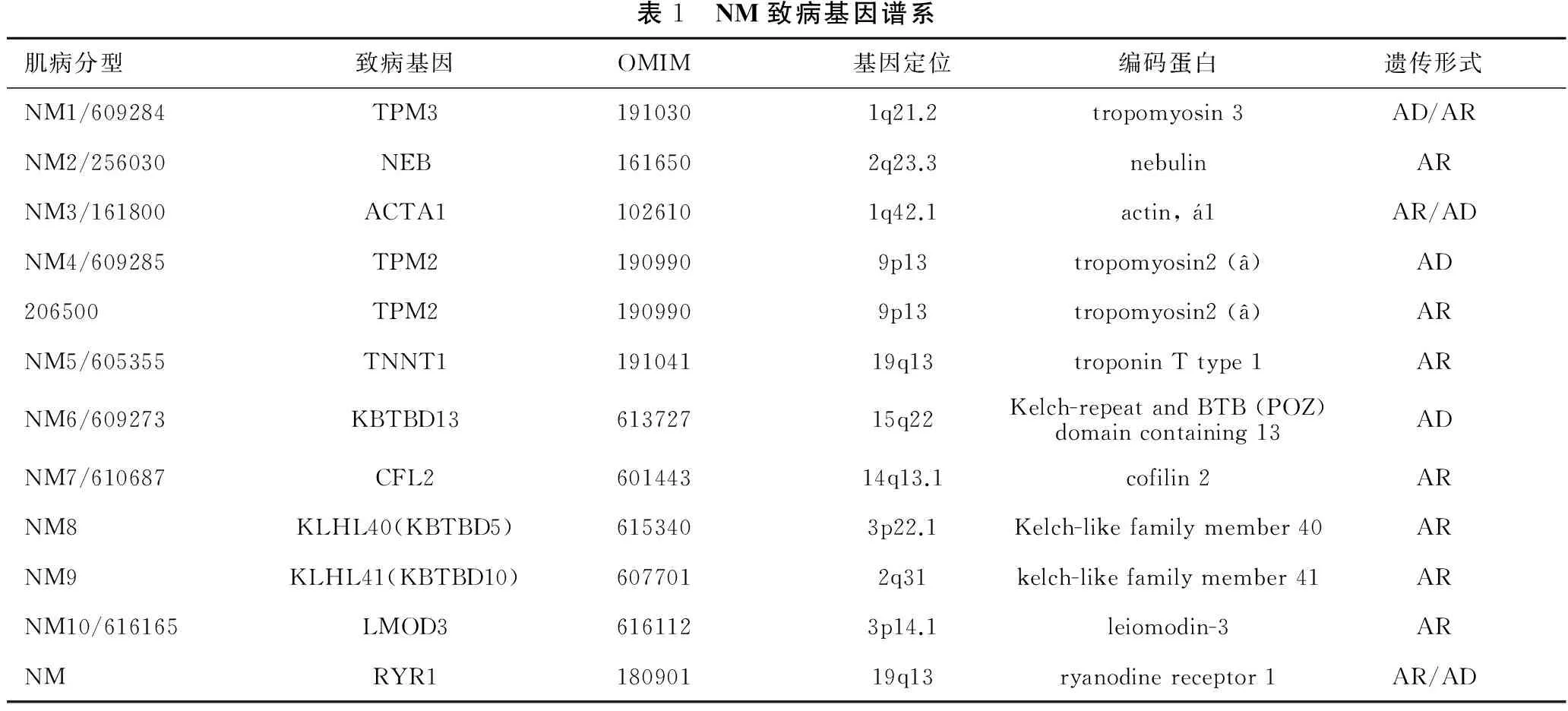
表1 NM致病基因谱系肌病分型致病基因OMIM基因定位编码蛋白遗传形式NM1/609284TPM31910301q21.2tropomyosin3AD/ARNM2/256030NEB1616502q23.3nebulinARNM3/161800ACTA11026101q42.1actin,1AR/ADNM4/609285TPM21909909p13tropomyosin2(â)AD206500TPM21909909p13tropomyosin2(â)ARNM5/605355TNNT119104119q13troponinTtype1ARNM6/609273KBTBD1361372715q22Kelch-repeatandBTB(POZ)domaincontaining13ADNM7/610687CFL260144314q13.1cofilin2ARNM8KLHL40(KBTBD5)6153403p22.1Kelch-likefamilymember40ARNM9KLHL41(KBTBD10)6077012q31kelch-likefamilymember41ARNM10/616165LMOD36161123p14.1leiomodin-3ARNMRYR118090119q13ryanodinereceptor1AR/AD
NEB变异是NM最常见原因,该基因拥有183个外显子,编码700 kDa的伴肌动蛋白。该蛋白为人类肌节中最大的蛋白之一,是细肌丝的组成部分,是肌动蛋白结合蛋白,具有高度重复序列结构,重复序列结构可以结合肌动蛋白。目前发现140个NEB突变,均呈AR遗传且多为复合杂合突变,未发现常见的突变或热点突变,大部分为移码突变及无义突变,可见错义突变、剪切突变。由于NEB巨大,以往的DNA测序使用传统的Sanger法,仅局限于测序伴肌动蛋白的C-末端(因该端与Z线锚定),如今下一代测序技术(NGS)为NEB变异的诊断带来了极大便利。如Scoto等[24]使用NGS报道NEB基因55号外显子4 bp重复的移码突变c.24372_24375dup (P.Val8126fs)。NEB还存在一些特殊突变,如1例德系犹太人于NEB基因的55号外显子及54、55号内含子部分区域被发现2502 bp缺失突变[30],为NEB分子生物学诊断带来了挑战。
ACTA1是第二常见的致病基因,编码骨骼肌a-肌动蛋白,该蛋白存在于Ⅰ型和Ⅱ型肌纤维中,约有25%的患者存在ACTA1基因突变[21]。目前已发现200余种ACTA突变,主要为错义突变,90%为AD遗传,10%为AR遗传。其中,约50%的患者表现为先天性NM严重型。ACTA1也是唯一一个与核内杆状体相关的基因,该基因变异亦引起斑马体肌病,也被认为是NM的一种,目前世界上仅报道了2例,骨骼肌病理不仅发现了斑马体,还显示了杆状体。
TPM2和TPM3分别编码β-原肌球蛋白和慢型α-原肌球蛋白,遗传方式为AR/AD,占5%以下的NM。迄今为止,已报道TPM2基因错义突变导致的NM:如Q147P[27]、E41K[9]、E117K[31];缺失突变:p.E138del[32]、p.E139del[9-10]。TPM3已报道为AD遗传的错义突变及AR遗传的1 bp的缺失突变。由于慢型α-原肌球蛋白仅存在于慢肌肌纤维中(Ⅰ型肌纤维),当肌细胞杆状体仅存在于Ⅰ型肌纤维时,可优先考虑TPM3变异。TNNT1和CFL2突变引起NM,目前报道的均为AR遗传。迄今为止,TNN1仅报道了1例无义突变,CFL2报道了几例纯合错义突变及缺失突变[19,23]。CFL2编码肌肉的肌动蛋白结合蛋白丝切因子-2,丝切因子-2缺乏可导致肌动蛋白解聚作用减少,导致其在肌纤维中的聚集形成杆状体[28]。KBTBD13发现了3个AD遗传的错义突变;KLHL40、KLHL41 和LMOD3均为AR遗传。目前报道了纯合/复合杂合突变,其中KLHL41移码突变可以导致严重的临床表现,新生儿即死亡;错义突变损害运动功能,患者可以存活至儿童晚期或成年早期[33]。
RYR1导致的单纯的NM非常少见,目前仅报道1例[34],该研究使用NGS发现1例2岁极严重NM患者的RYR1第33号外显子c.4718C>T (p.1573Pro>Leu)及47号外显子c.7585G>A (p.2529Asp>Asn) 复合杂合突变。RYR1变异报道较多的是轴空/杆状体肌病[18, 34-35]。轴空/杆状体肌病在NM中非常少见,已报道致病基因除了最多见的RYR1外还有NEB[22]和ACTA1[36]。帽状体肌病目前已知的致病基因为ACTA1[14]、TPM2[9, 11]、TPM3[37-38]。
4 总结
随着分子生物学技术的发展与应用,越来越多的NM病例被报道,人们认识了更多的NM的临床、病理及遗传学特征,因此,“杆状体”不能再作为诊断本组肌病的金标准,而是必须结合分子生物学诊断。同时,分子生物学研究的进展也推动了人们对该病发病机制的探索,随着动物模型的建立,NM的发病机制研究以及基因治疗将成为新的研究热点。
[1] Colombo I, Scoto M, Manzur AY, et al. Neurology, 2015, 84: 28.
[2] North KN, Wang CH, Clarke N, et al. NMD, 2014, 24: 97.
[3] Lehtokari VL, Pelin K, Herczegfalvi A, et al. NMD, 2011, 21: 556.
[4] Wallgren-Pettersson C, Laing NG. NMD, 2000, 10: 299.
[5] Ryan MM, Schnell C, Strickland CD, et al. Ann Neurol, 2001, 50: 312.
[6] Ravenscroft G, Miyatake S, Lehtokari VL, et al. Am J Hum Genet, 2013, 93: 6.
[7] Jungbluth H, Sewry CA, Counsell S, et al. NMD, 2004, 14: 779.
[8] Quijano-Roy S, Avila-Smirnow D, Carlier RY. NMD, 2012, 22: 68.
[9] Tajsharghi HOM, Lindberg C, Oldfors A. Arch Neurol, 2007, 64: 1334.
[10] 尹西, 蒲传强, 黄旭升, 等.中华神经科杂志, 2013, 46: 676.
[11] Clarke NF, Domazetovska A, Waddell L, et al. NMD, 2009, 19: 348.
[12] de Paula AM, Franques J, Fernandez C, et al. NMD, 2009, 19: 685.
[13] Ohlsson M, Fidzianska A, Tajsharghi H, et al. Neurology, 2009, 72: 1961.
[14] Hung RM, Yoon G, Hawkins CE, et al. NMD, 2010, 20: 238.
[15] Waddell LB, Kreissl M, Kornberg A, et al. NMD, 2010, 20: 464.
[16] Malfatti E, Schaeffer U, Chapon F, et al. NMD, 2013, 23: 992.
[17] Ohlsson M, Quijano-Roy S, Darin N, et al. Neurology, 2008, 71: 1896.
[18] Scacheri P, Hoffman E, Fratkin J, et al. Neurology, 2000, 55: 1689.
[19] Hernandez-Lain A, Husson I, Monnier N, et al. Eur J Med Genet, 2011, 54: 29.
[20] Wallgren-Pettersson C, Sewry CA, Nowak KJ, et al. Semin Pediatr Neurol, 2011, 18: 230.
[21] de Sanctis JT, Cumbo-Nacheli G, Dobbie D, et al. AIDS Read, 2008, 18: 90.
[22] Romero NB, Lehtokari VL, Quijano-Roy S, et al. Neurology, 2009, 73: 1159.
[23] Wattanasirichaigoon D, Swoboda K, Takada F, et al. Neurology, 2002, 59: 613.
[24] Scoto M, Cullup T, Cirak S, et al. Euro J Hum Genet, 2013, 21: 1249.
[25] Ravenscroft G, Wilmshurst JM, Pillay K, et al. NMD, 2011, 21: 31.
[26] Donner K, Ollikainen M, Ridanpää M, et al. NMD, 2002, 12: 151.
[27] Johnston JJ, Kelley RI, Crawford TO, et al. Am J Hum Genet, 2000, 67: 814.
[28] Agrawal PB, Greenleaf RS, Tomczak KK, et al. Am J Hum Genet, 2007, 80: 162.
[29] Dhanoa BS, Cogliati T, Satish AG, et al. Hum Genet, 2013, 7: 13.
[30] Anderson SL, Ekstein J, Donnelly MC, et al. Human Genet, 2004, 115: 185.
[31] Karpicheva OE, Robinson P, Piers A, et al. Arch Biochem Biophys, 2013, 536: 25.
[32] Tasca G, Fattori F, Ricci E, et al. Acta Neuropathol, 2013, 125: 169.
[33] Gupta-Vandana A, Ravenscroft G, Shaheen R, et al. Am J Hum Genet, 2013, 93: 1108.
[34] Kondo E, Nishimura T, Kosho T, et al. Am J Hum Genet, 2012, 158: 772.
[35] Davis M, Haan E, Jungbluth H, et al. NMD, 2003, 13: 151.
[36] Jungblutha H, Browna SC, Nowak KJ, et al. NMD, 2001, 11: 35.
[37] Schreckenbach T, Schroder JM, Voit T, et al. NMD, 2014, 24: 117.
[38] Fidzianska A, Madej-Pilarczyk A, Hausmanowa-Petrusewicz I. Clin Neuropathol, 2014, 33: 61.
R746
A
1004-1648(2017)05-0383-03
国家自然科学基金青年基金(81300977);国家自然科学基金面上项目(81671113)
210008南京大学医学院附属鼓楼医院神经内科(赵燕,刘卓,常蕾蕾,卢正娟,吴家勇,徐运),病理科(牛丰南)
徐运
2016-12-10
2016-12-20)
猜你喜欢
广西医科大学学报(2022年5期)2022-06-07
昆明医科大学学报(2022年3期)2022-04-19
今日农业(2021年5期)2021-11-27
现代畜牧科技(2021年5期)2021-07-20
中国畜牧杂志(2020年1期)2020-01-16
中南医学科学杂志(2019年6期)2019-12-05
兽医导刊(2019年24期)2019-02-12
浙江农业科学(2016年11期)2016-05-04
中国医科大学学报(2015年10期)2015-03-01
中国当代医药(2015年26期)2015-03-01
