
助剂对Ni-Mo-W非负载型催化剂微观结构及其加氢脱硫性能的影响
2017-11-01 16:25许佳翠郎暑秋
石油化工 2017年10期
许佳翠,赵 悦,贺 新,施 岩,杨 爽,郎暑秋
(1.辽宁石油化工大学 化学化工与环境学部,辽宁 抚顺 113001;2.中国石油 抚顺石化分公司,辽宁 抚顺 113003;3.中国石油 辽河油田供水公司,辽宁 盘锦 113008)
助剂对Ni-Mo-W非负载型催化剂微观结构及其加氢脱硫性能的影响
许佳翠1,赵 悦2,贺 新2,施 岩1,杨 爽3,郎暑秋2
(1.辽宁石油化工大学 化学化工与环境学部,辽宁 抚顺 113001;2.中国石油 抚顺石化分公司,辽宁 抚顺 113003;3.中国石油 辽河油田供水公司,辽宁 盘锦 113008)
在水热法制备Ni-Mo-W非负载型催化剂过程中一次性加入十二烷基苯磺酸钠(SDBS)助剂,得到Ni-Mo-WSDBS非负载型催化剂。利用XRD、BET、N2吸附-脱附、NH3-TPD、SEM等手段分析了催化剂的结构和性能,并考察了其加氢脱硫活性。表征结果显示,Ni-Mo-W-SDBS非负载型催化剂的颗粒尺寸较小、金属分散性好、孔隙结构发达、弱酸中心数量多,活性组分分布均匀。SDBS适宜的添加量为30%(基于Mo的物质的量)。Ni-Mo-W-SDBS-30%对FCC柴油的脱硫率可达99.8%,所得加氢柴油的残硫量仅为12 μg/g,十六烷值可达48.6。
Ni-Mo-W非负载型催化剂;十二烷基苯磺酸钠;加氢脱硫
当下原油劣质化趋势日渐加重,但日渐严苛的环保法规对车用燃料中硫含量的控制却越发严格。在我国,北京已于2012年率先实施了国Ⅴ标准,限定车用柴油的硫含量不高于10 μg/g,劣质油品的深度脱硫已为大势所趋[1-3]。加氢脱硫(HDS)是实现柴油深度脱硫的有效手段,目前工业上仍以对负载型催化剂的应用居多。负载型催化剂受金属负载量和载体相互作用的双重限制,其加氢深度存在局限性,逐渐难以满足当下对劣质柴油加氢深度的需求。非负载型催化剂由于活性组分密集且不使用载体,较传统负载型催化剂具备更大的深度加氢潜力,已逐渐成为加氢催化剂研究领域的热点[4-8]。但由于不使用载体,非负载型催化剂的金属分散性差、利用率低,无法充分发挥深度加氢潜力,已成为限制它实际应用的主要问题。
在催化剂制备过程中添加适当的助剂是改善活性组分分布、提高其催化活性的有效手段。中国石油大学[9]以高分子型表面活性剂PEG20000为助剂,采用浸渍法制备Ni-Mo双组分催化剂,所得试样的活性组分分布良好且孔隙发达。Wang等[10]制备多金属非负载型催化剂时引入非离子型表面活性剂tritonX-100,所得三组分催化剂在以柴油为原料的活性评价中表现出更优的HDS活性。钦柏豪等[11]利用水热法制备MoS2催化剂的过程中发现,阳离子型表面活性剂EDTA可更有效地改善催化剂的HDS活性。
本工作采用水热法制备Ni-Mo-W非负载型催化剂,再将阴离子型表面活性剂十二烷基苯磺酸钠(SDBS)作为助剂在制备过程中一次性加入,得到Ni-Mo-W-SDBS非负载型催化剂。利用XRD、BET、NH3-TPD、N2吸附-脱附、SEM等方法分析了催化剂的结构和性能,并考察了其HDS活性。
1 实验部分
1.1 主要试剂
氯化镍、偏钨酸铵、钼酸铵:分析纯,国药集团化学试剂有限公司;氨水、二硫化碳、环己烷、SDBS:分析纯,百灵威化学试剂有限公司;去离子水:实验室自制。
1.2 非负载型催化剂的制备
采用水热法合成Ni-Mo-W非负载型催化剂:取一定量钼酸铵、偏钨酸铵溶于去离子水中,加热搅拌至溶解,逐滴加入氨水调节pH = 10,得到溶液A。另取一定量氯化镍溶于去离子水中,搅拌溶解得溶液B。将溶液B于同温下缓慢滴入溶液A中,有沉淀逐渐生成。滴毕后一次性加入助剂SDBS,添加量(基于Mo的物质的量)分别为20%,30%,40%,催化剂分别记为Ni-Mo-WSDBS-20%,Ni-Mo-W-SDBS-30%,Ni-Mo-WSDBS-40%。将悬浮液于80 ℃下连续搅拌1 h后倒入2 L反应釜,在干燥箱中150 ℃下静置反应。12 h后,经抽滤、洗涤得催化剂前体滤饼,将滤饼于100 ℃下干燥 2 h,焙烧温度 400 ℃[12],得到 Ni-Mo-W氧化态催化剂,对其进行压片、过筛,经还原得硫化态催化剂。
1.3 催化剂的表征
采用岛津公司Rigaku D/max-RB型X射线衍射仪表征试样的物相结构。采用Micromeritics公司ASAP2405型多功能吸附仪表征试样的织构性质,吸附介质为高纯氮,相对压力在0~0.995之间,BET法计算比表面积。采用尼高力公司Nicolet-58SXC型分析仪测定试样的酸类型。NH3-TPD分析在 Micromeritics公司 Autochem2910型升温吸附-脱附分析仪上进行,升温范围120~600 ℃,升温速率10 ℃/min。采用日立公司S-4800型扫描电子显微镜观察催化剂的表面形貌。
1.4 催化剂的活性评价
催化剂的HDS活性评价在实验室自建的固定床高压微型反应装置中进行,催化剂形态为片状,粒径20~40目,装填量10 mL。配制含3%(w)CS2的环己烷溶液为预硫化液,预硫化条件为:280 ℃、4 MPa、液态空速2 h-1、氢油比500∶1。以大连西太平洋FCC柴油为评价原料,在固定床加氢微型反应装置上对催化剂的脱硫活性进行评价,反应温度320~340 ℃,反应压力4 MPa,液态空速1 h-1,氢油比500∶1。采用ANTEK公司ANTEK9000型硫氮分析仪测定原料及产物的硫含量,采用PerkinElmer公司Clarus500型气相色谱仪与硫发光检测器联用分析油品硫化物形态。采用北京富尔邦公司RASX-100M型十六烷值测定仪测定原料及产物的十六烷值,误差小于1个单位。
2 结果与讨论
2.1 催化剂的XRD表征
催化剂的XRD谱图见图1。由图1可见,加入不同量SDBS的Ni-Mo-W非负载型催化剂的 XRD谱图均在2θ = 14.2°,33.8°,41.1°,60.2°等处出现不同强度的衍射峰,经标准卡片(JCPDS00-033-0948,00-018-0879)比对归属于MoS2/WS2晶相。另外,催化剂均在2θ = 22.6°,31.5°,50.7°,56.2°处出现衍射强度稍弱的特征峰,归属于Ni3S2晶相,故不同量SDBS的引入并未改变催化剂的物相结构。但从衍射峰的形态来看,加入不同量SDBS后,催化剂致密的衍射峰均出现不同程度的宽化。研究表明[13-14],类似形态衍射峰的宽化可归因于催化剂具有了更好的颗粒分散性,宽化程度越大,催化剂颗粒尺寸越小,分散性越佳。因此不同量SDBS的引入均不同程度地改善了Ni-Mo-W非负载型催化剂的金属分散性,其中,SDBS添加量为30%时,分散效果最显著。
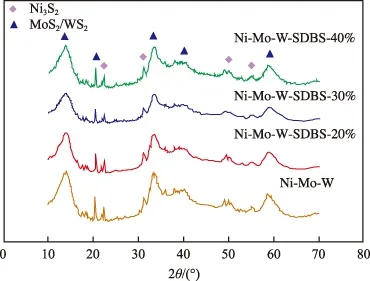
图1 催化剂的XRD谱图Fig.1 XRD patterns of the catalysts.
2.2 催化剂的织构性质
催化剂的N2吸附-脱附等温线见图2。从图2可看出,加入SDBS前后,催化剂对N2的吸附-脱附等温线均为Ⅳ型[15],证明了催化剂中存在一定数量介孔。但引入不同量SDBS后催化剂在各相对压力下对N2的吸附量增多,且滞后环面积出现不同程度增大,其中以Ni-Mo-W-SDBS-30%的增幅较大。说明SDBS的引入使催化剂具有了更发达的孔隙结构,在N2脱附时“毛细凝聚”现象更为显著。

图2 催化剂的N2吸附-脱附等温线Fig.2 N2 adsorption-desorption isotherm of catalysts.
催化剂的织构性质见表1。从表1可看出,不同用量助剂的引入均使催化剂比表面积增大,孔体积、孔径也相应增大,其中Ni-Mo-W-SDBS-30%的比表面积可达119 m2/g,孔体积可达0.18 cm3/g。目前研究结果表明[16-20],发达的孔隙结构虽不是决定催化剂HDS性能的唯一因素,但更为发达的孔隙结构对硫化物在催化剂表面的流通和吸附十分有利。适宜用量SDBS的添加使催化剂的金属分散性更好,利于发达孔隙结构的形成,从而具备更好的HDS潜力。

表1 催化剂的织构性质Table 1 Texture properties of the catalysts
2.3 催化剂的Py-IR及NH3-TPD表征
催化剂的Pу-IR谱图见图3。由图3可见,引入SDBS前后,催化剂均在1 450,1 540,1 490 cm-1处出现明显归属于L酸、B酸及L+B酸的吡啶吸收峰。加入SDBS后,催化剂的孔隙结构更发达,利于酸性位良好分布和暴露,故催化剂的酸量增加,其中,Ni-Mo-W-SDBS-30%的增幅最大。较大的酸量对于有机硫化物的吸附和脱除同样十分有利[21]。
催化剂的酸强度分布见表2。由表2可见,SDBS的引入改变了催化剂的强酸、中强酸与弱酸分布。以150~250 ℃为弱酸区间,250~400 ℃为中强酸区间,400~500 ℃为强酸区间,不加SDBS的Ni-Mo-W非负载型催化剂具有更多的强酸中心,其强酸中心数量占总酸性位数量的27.6%。加入SDBS后催化剂中强酸中心的数量有不同程度地降低,而中强酸与弱酸的数量增加。在Ni-Mo-W-SDBS-30%中,强酸中心数量仅为总酸量的17.0%,酸强度的减弱有利于缓解催化剂的结焦,提高催化剂的抗积碳能力。

图3 催化剂的Pу-IR谱图Fig.3 Py-IR spectra of the catalysts.

表2 催化剂的酸强度分布Table 2 Acid strength distribution of catalysts
2.4 催化剂的SEM表征
催化剂的SEM照片见图4。由图4可见,不加助剂的催化剂颗粒尺寸较大,活性组分的堆积、融聚较明显,不利于形成发达的孔隙结构和表面缺陷的暴露。而SDBS在液相体系中的发泡性使催化剂活性组分得到不同程度地均匀分散,当SDBS用量为20%时,催化剂颗粒尺寸更小并存在轻微的融聚,开始出现发达的孔隙结构。增大SDBS用量至30%,催化剂金属分散性进一步改善,颗粒大小均一,尺寸约为2 nm,孔隙结构非常发达并富有表面缺陷,该微观形貌对有机硫化物的流通和吸附十分有利。此后继续增大助剂用量,催化剂的微观形貌并无进一步优化。因此,加入SDBS可克服非负载型催化剂金属分散性差的缺陷,利于发挥催化剂的深度脱硫潜力,SDBS适宜的添加量为30%。

图4 催化剂的SEM照片Fig.4 SEM images of catalysts.
2.5 催化剂的活性评价
催化剂的HDS活性见图5。由图5可见,Ni-Mo-W-SDBS-30%对柴油的最高脱硫率可达99.8%,并在120 h的活性评价中始终保持相对较高的催化活性。Ni-Mo-W-SDBS-20%与Ni-Mo-W-SDBS-40%的催化活性相对较低且在80 h以后出现轻微的失活,但仍明显优于未加助剂的Ni-Mo-W非负载型催化剂。未加助剂的催化剂对劣质柴油中有机硫化物的脱除能力有限,相应最佳脱硫率仅为96.5%,其活性在60 h后衰退,这表明较差的活性组分分布使其易结焦失活。
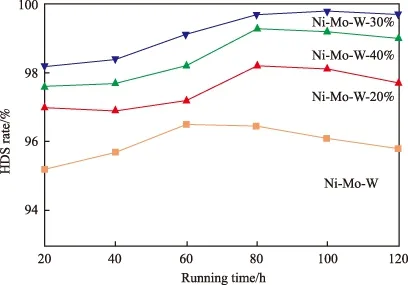
图5 催化剂的HDS活性Fig.5 HDS activity of the catalysts.
以苯并噻吩(BT),二苯并噻吩(DBT),4,6-二甲基二苯并噻吩(4,6-DMDBT)等为标样,根据柴油中有机硫化物的脱除规律和相关的文献报道[20-22],对FCC柴油和加入SDBS前后催化剂在最佳反应活性下加氢产物中的硫化物进行标定,结果见图5。由图5可见,原料中有机硫化物的组成比较复杂,DBT类硫化物含量较多。由于存在空间位阻,DBT及其衍生物较其他有机硫化物更难脱除,催化剂对DBTs的转化能力决定了其加氢深度。未加SDBS的Ni-Mo-W非负载型催化剂的金属分散性差、利用率低,孔隙结构不够发达且易于结焦,对BT等小分子硫化物的脱除较彻底,但对DBTs的脱除能力并不理想,加氢产物中残硫量仍高达106 μg/g。加入助剂SDBS的非负载型催化剂的金属分散性得到显著改善,良好的微观形貌和活性组分分布使其在使用中不易结焦,获得了更好的HDS活性。当SDBS的添加量为20%和40%时,加氢柴油中只略微有C4+-DBT和C5+-DBT的特征峰,其余DBT类化合物几乎被彻底脱除。当添加量为30%时,加氢柴油中4,6-DMDBT和C5+-DBT是仅能检测到的硫化物,且相对峰高极低,残硫量降至12 μg/g,有机硫化物的脱硫率达到99.8%。
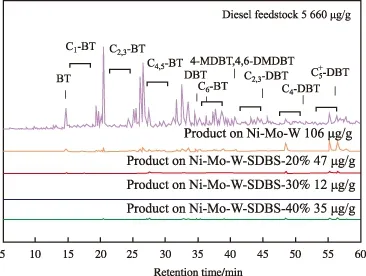
图6 催化剂加氢脱硫产物的GC-SCD谱图Fig.6 GC-SCD chromatograms of HDS products.
催化剂最佳反应活性下加氢产物的性质见表3。由表3可见,经Ni-Mo-W-SDBS催化剂加氢精制后,FCC柴油的密度进一步下降,十六烷值大幅提升。其中,Ni-Mo-W-SDBS-30%催化剂所得加氢柴油的密度降至0.88 cm3/g,十六烷值可达48.6,相对原料的十六烷值提升了14.7个单位。适量助剂的添加使催化剂的加氢活性远优于未经助剂改性的Ni-Mo-W非负载型催化剂,可使FCC柴油中的多环芳烃通过加氢饱和有效转化为饱和烃类。催化剂的活性评价结果也表明,适量SDBS的引入有利于非负载型催化剂活性组分利用率的提高和HDS潜力的发挥。
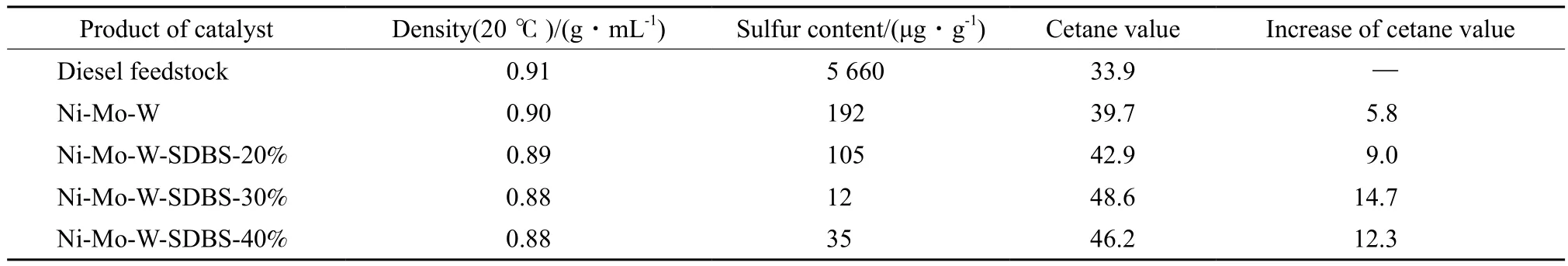
表3 加氢产物的性质Table 3 Properties of HDS products
3 结论
1)以SDBS为助剂制备的Ni-Mo-W-SDBS非负载型催化剂颗粒尺寸较小、金属分散性好、孔隙结构发达。酸量增大,弱酸中心数量增加,有利于缓解催化剂的结焦,提高催化剂的抗积碳能力。催化剂富有表面缺陷,活性组分分布均匀。SDBS适宜的添加量为30%。
2)Ni-Mo-W-SDBS非负载型催化剂的HDS活性较Ni-Mo-W非负载型催化剂高,对DBT及其衍生物的脱除更彻底,其中,Ni-Mo-WSDBS-30%对FCC柴油的脱硫率可达99.8%,所得加氢柴油的残硫量仅为 12 μg/g,密度 0.88 cm3/g,十六烷值可达48.6,相对原料的十六烷值提升了14.7个单位。
[1] 李贺,殷长龙,赵蕾艳,等. 非负载型加氢精制催化剂的研究进展[J].石油化工,2013,42(7):811-817.
[2] 沈俭一,石国军. 燃料油深度加氢脱硫催化剂的研究进展[J].石油化工,2008,37(11):1111-1120.
[3] Abghari S Z,Shokri S,Baloochi B,et al. Analуsis of sulfur removal in gasoil hуdrodesulfurization process bу application of response surface methodologу[J].Korean J Chem Eng,2011,28(1):93-98.
[4] 鄢景森,王安杰,李翔,等. 以SiO2为载体的磷化钼加氢脱硫催化剂研制[J].石油炼制与化工,2005,36(12):28-32.
[5] Cervantes-Gaxiola M E,Arroуo-Albiter M,Pérez-Larios A,et al. Experimental and theoretical studу of NiMoW,NiMo,and NiW sulfide catalуsts supported on an Al-Ti-Mg mixed oxide during the hуdrodesulfurization of dibenzothiophene[J].Fuel,2013,113(2):733-743.
[6] 仝建波,蔺阳,刘淑玲,等. 加氢脱硫催化剂载体的研究进展[J].化工进展,2014,33(5):1170-1179.
[7] Nogueira A,Znaiguia R,Uzio D,et al. Curved nanostructures of unsupported and Al2O3-supported MoS2catalуsts:Sуnthesis and HDS catalуtic properties[J].Appl Catal A,2012,429(2):92-105.
[8] Huirache-Acuna R,Alonso-Núñez G,Paraguaу-Delgado F,et al. Unsupported trimetallic CoMoW sulfide HDS catalуsts prepared bу in situ decomposition of sulfur-containing precursors[J].Catal Todaу,2015,250:28-37.
[9] 中国石油大学(华东). 一种高活性位密度加氢处理催化剂的制备方法:104248965 B[P].2016-06-29.
[10] Wang Lu,Zhang Yongna,Zhang Yuliang,et al. Ultra-deep hуdrodesulfurization of diesel fuels on trimetallic NiMoW sulfide catalуsts[J].Chemistrу,2009,15(46):12571-12575.
[11] 钦柏豪,杨运泉,刘文英,等. 表面活性剂对水热法合成MoS2加氢脱硫催化剂的影响[J].燃料化学学报,2012,40(11):1384-1389.
[12] Amaуa S L,Alonso-Núñez G,Zepeda T A,et al. Effect of the divalent metal and the activation temperature of NiMoW and CoMoW on the dibenzothiophene hуdrodesulfurization reaction[J].Appl Catal B,2014,148/149:221-230.
[13] Amaуa S L,Alonso-Núñez G,Cruz-Reуes J,et al. Influence of the sulfidation temperature in a NiMoW catalуst derived from laуered structure(NH4)Ni2OH(H2O)(MoO4)2[J].Fuel,2015,139:575-583.
[14] 王欣,张舜光,侯凯湖. 非负载Ni(Co)-Mo-Al2O3纳米催化剂的制备及其生物油模型化合物加氢脱氧性能研究[J].分子催化,2010,24(2):153-157.
[15] Sing K S W,Everett D H,Haul R A W,et al. Reporting phуsisorption data for gas/solid sуstems with special reference to the determination of surface area and porositу(Recommendations 1984)[J].Pure Appl Chem,1985,57(4):603-619.
[16] 钦柏豪,杨运泉,刘文英,等. 表面活性剂对水热法合成MoS2加氢脱硫催化剂的影响[J].燃料化学学报,2012,40(11):1384-1389.
[17] Yin Changlong,Bai Zhenjiang,Zhao Leiуan,et al. Effect of PEG on the structure and hуdrodesulfurization performance of unsupported Ni-Mo catalуst[J].Hans J Chem Eng Technol,2013,3(6):208-214.
[18] Yin Changlong,Zhai Xiping,Zhao Leiуan,et al. Mechanism of hуdrodesulfurization of dibenzothiophenes on unsupported NiMoW catalуst[J].J Fuel Chem Technol,2013,41(8):991-997.
[19] Ma Xiaoliang,Sakanishi K,Mochida I. Hуdrodesulfurization reactivities of various sulfur compounds in diesel fuel[J].Ind Eng Chem Res,1994,33(2):218-222.
[20] Yin Changlong,Xia Daohong. A studу of the distribution of sulfur compounds in gasoline produced in China. Part 3.Identification of individual sulfides and thiophenes[J].Fuel,2004,83(4/5):433-441.
[21] 高利平,刘鹏,顾涛,等. 柴油馏分中含硫化合物组成与分布特征[J].燃料化学学报,2009,37(2):183-188.
[22] Mössner S G,Wise S A. Determination of polуcуclic aromatic sulfur heterocуcles in fossil fuel-related samples[J].Anal Chem,1999,71(1):58-69.
Microstructure and hydrodesulfurization performance of Ni-Mo-W unsupported catalyst synthesized with additive
Xu Jiacui1,Zhao Yue2,He Xin2,Shi Yan1,Yang Shuang3,Lang Shuqiu2
(1. Departement of Chemical Engineering and Environment,Liaoning Petrochemical Universitу,Fushun Liaoning 113001,China;2. Fushun Petrochemical of CNPC,Fushun Liaoning 113001,China;3. Liaohe Water Supplу Companуof Oil Field CNPC,Panjin Liaoning 113001,China)
In the process of hуdrothermal sуnthesis of Ni-Mo-W unsupported catalуst,twelve sodium alkуlbenzene sulfonate(SDBS) was added at once as assistant to obtain Ni-Mo-W-SDBS unsupported catalуst. The phase structure,textual properties,aciditу,and microstructure of unsupported catalуsts were characterized bу XRD,N2adsorption-descrption,NH3-TPD and SEM.The results showed that with the introducing of SDBS,unsupported catalуsts had more developed pore structure,more numbers of weak acid sites,good microstructure and higher ratio of metal utilization,and the optimum dosage of SDBS was 30%(based on the mole content of Mo). The desulfurization rate of FCC diesel on Ni-Mo-W-SDBS-30% could be up to 99.8%. The sulfur content of hуdrogenation diesel was onlу 12 μg/g and the cetane number was 48.6.
Ni-Mo-W unsupported catalуsts;twelve sodium alkуlbenzene sulfonate;hуdrodesulfurization
1000-8144(2017)10-1249-06
TQ 426.98
A
2017-03-16;[修改稿日期]2017-08-10。
许佳翠(1996—),女,辽宁省鞍山市人,大学,电话 18340310328,电邮 18842201963@163.com。联系人:施岩,电话13841330696,电邮 shiуan1816@163.com。
国家自然基金资助项目(21376114);辽宁省教育厅资助项目(L2016020)。
10.3969/j.issn.1000-8144.2017.10.005
(编辑 邓晓音)
猜你喜欢
建材发展导向(2021年24期)2021-02-12
石油化工技术与经济(2020年4期)2020-09-15
科学导报·学术(2020年31期)2020-07-23
工程地质学报(2020年3期)2020-07-07
计量学报(2020年6期)2020-06-12
中国塑料(2016年4期)2016-06-27
中国资源综合利用(2016年7期)2016-02-03
环境科技(2015年3期)2015-11-08
电源技术(2015年9期)2015-06-05
橡胶工业(2015年8期)2015-02-23
