
氮掺杂还原氧化石墨烯与吡啶共聚g-C3N4复合光催化剂及其增强的产氢活性
2017-11-01 17:29程若霖金锡雄樊向前田建建张玲霞施剑林
物理化学学报 2017年7期
程若霖 金锡雄 樊向前 王 敏 田建建 张玲霞,* 施剑林,*
(1中国科学院上海硅酸盐研究所,高性能陶瓷和超微结构国家重点实验室,上海 200050;2中国科学院大学,北京 100049)
氮掺杂还原氧化石墨烯与吡啶共聚g-C3N4复合光催化剂及其增强的产氢活性
程若霖1,2金锡雄1,2樊向前1,2王 敏1,2田建建1,2张玲霞1,2,*施剑林1,2,*
(1中国科学院上海硅酸盐研究所,高性能陶瓷和超微结构国家重点实验室,上海 200050;2中国科学院大学,北京 100049)
采用一种新颖有效的席夫碱化学法合成吡啶共聚改性的g-C3N4,其可见光催化产氢性能较(由尿素为前驱物制备的)纯g-C3N4显著增强。在此基础上,又进一步通过一步煅烧的方法构建了吡啶改性g-C3N4和N掺杂还原氧化石墨烯(N-rGO)的复合物,其产氢活性得到了进一步地提高,氢气产量最高达到304µmol·h−1,分别为纯g-C3N4和吡啶改性g-C3N4的11.7倍和3.1倍。除了其增强的可见光吸收能力,增大的表面积,我们认为:吡啶环作为分子内电子受体,N-rGO作为“电子转移活性位”,二者共同促进了光生载流子分离和转移,从而显著增强了该复合体系的光催化活性。
光催化;石墨相氮化碳;共聚合;分解水制氢;纳米复合材料
1 Introduction
The ever-increasing energy demands and environment crisis are among the most serious global problems. Artificial photosynthesis, as a promising solution typically by producing chemical fuels (H2, CH4, CH3OH, etc.) via splitting water and reducing CO2, has been being a key focus but remains a great challenge for researchers. Traditional photocatalysts (e.g. TiO2)with wide band gap and low solar-energy utilization efficiency cannot satisfy the requirements of practical applications1.Persistent exploration of efficient visible-light-driven photocatalysts has developed various strategies to modify semiconductors, including band engineering by heterogeneous doping, sensitizing by incorporating efficient visible-light converters (e.g. metal-organic dyes) and discovering new narrow band-gap semiconductors, etc2,3.
Conjugated polymer semiconductors show a number of advantages in solar energy conversion such as simple manufacture, low cost, lightweight, flexibility, etc4,5. Graphitic carbon nitride (g-C3N4), which has graphene-analogue two-dimensional (2D) structure, has been introduced as a metal-free photocatalyst for highly efficient H2evolution under visible light6,7. Featured with facile preparation, high physicochemical stability and an appealing electronic structure combined with a medium band gap (2.7 eV), g-C3N4has become a promising candidate of visible light photocatalyst.Nevertheless, the photocatalytic performance of pure g-C3N4is still far from satisfaction because of the still low utilization of visible light and the fast recombination of photogenerated charge carriers. Several strategies, such as constructing nanostructures and nanocomposites8,9, doping heterogeneous elements/units10,11, and copolymerization with other molecular fragments12, have been exploited to optimize the electronic structure of g-C3N4and enhance its photocatalytic activity and efficiency. Among them, copolymerization is an effective route to modify g-C3N4with a number of aromatic rings, which can be incorporated into g-C3N4networks to broaden the range of visible light response and improve the charge carrier separation13. Though various organic monomers with cyano and amino groups have been selected as the copolymerization blocks, they are usually of high cost and have a limited menu of precursor choices. Inspired by the Schiff-base chemistry, our group has confirmed the possibility of using a series of aromatic aldehydes as co-monomers to construct novel g-C3N4-based photocatalysts by copolymerization or post-grafting method14,15. Typically, the aromatic rings are connected by adjacent 3,s-triazine units instead of being incorporated into the inner 3,s-triazine units.
Another common methodology is to construct heterojunction interfaces between g-C3N4and other semiconductors16−18. As graphene and g-C3N4have similar 2D network and sp2conjugated π structure, it is easy to predict that the introduction of graphene into g-C3N4will result in excellent electronic properties, optimized light absorbance and enhanced carrier mobility19. As reported, the construction of 2D layered junction between reduced graphene oxide (rGO) and g-C3N4can increase the contact area, which facilitates the charge transfer across the interface bond to graphene to join the coordinate redox reactions20and the separation of photogenerated electrons and holes21,22. Though graphene oxide (GO) is one of the main precursors to prepare graphene-based photocatalyst23and to modify g-C3N424, the presence of oxygen in its networks may cause the great decrease of electronic conductivity25.Melamine26, urea27and other precursors28,29of g-C3N4were used to couple with GO to construct the hybrid of rGO and g-C3N4for improved photocatalytic performance. However,these coupling processes commonly need an inert or reductive atmosphere to achieve rGO30and N-doped rGO (N-rGO)31at high temperature.
Herein, in striving to further elevate the catalytic efficiency of g-C3N4based photocatalyst, we integrated the advantages of copolymerization and composite construction into one system and fabricated a pyridine-modified g-C3N4/N-rGO composite by simple one-pot calcination. N-rGO has been incorporated in g-C3N4without the protection by inert or reductive atmosphere.The obtained catalysts showed extraordinarily high photocatalytic activity in water splitting to produce H2under visible light irradiation.
2 Experimental and computational section
2.1 Catalyst synthesis
2.1.1 Chemicals and reagents
Urea (> 99%) was obtained from Sinopharm Chemical Reagent Co., Ltd.; Triethanolamine (> 97%) and chloroplatinic acid hexahydrate (H2PtCl6) (> 37.5%, Pt basis) were purchased from Sigma-Aldrich Co. LLC.; Graphene oxide (GO, > 98%)was bought from XianFeng, Ltd.; 2,6-Pyridinedicarboxaldehyde(> 98%) was purchased from TCI Co., Ltd. All the chemicals were used as received.
2.1.2 Synthesis of pyridine-modified g-C3N4
In a typical synthesis procedure, 20 g urea and a certain amount of 2,6-pyridinedicarboxaldehyde were mixed thoroughly and then the mixture was put in an alumina crucible with a cover and heated to 550 °C for 2 h at a heating rate of 5 °C ·min−1. After undergoing various reactions at high temperature, as-constructed copolymer based on g-C3N4and pyridine were obtained, named as CN-x (x = 1, 3, 5, 10, 20 mg,2,6-pyridinedicarboxaldehyde), while pure g-C3N4was named as CN.
2.1.3 Fabrication of pyridine-modified g-C3N4/N-rGO composites
A one-pot calcination method was used here to synthesize pyridine modified g-C3N4/rGO composites with different mass fraction. A typical process is as follows: 20 g urea, 3 mg 2,6-pyridinedicarboxaldehyde and a different amount of GO were mixed homogeneously by grounding in an agate mortar,then the powder was put into an alumina crucible and calcined for 2 h at 550 °C. The resultant material was denoted as yGO-CN-3 (y = 1, 3, 5, 7, 10 mg, GO).
2.2 Characterization
X-ray diffraction measurements were conduct on a Rigaku D/Max 2200PC diffractometer with Cu Kαradiation (λ =15.418 nm) in the 2θ range of 10°−70°. Fourier transformed infrared (FTIR) spectra were collected using a Nicolet iS10 FTIR spectrometer. Transmission electron microscopic (TEM)images were obtained on a JEOL 200CX electron microscope operated at 200 kV. Nitrogen adsorptiondesorption isotherms at 77 K were carried out on a Micromeritics TriStar 3000 instrument. Before the measurement, all the samples were degassed at 150 °C for 8 h under flowing N2. X-ray photoelectron spectroscopy (XPS) measurement was conduct on a Thermo Scientific ESCALAB 250 spectrometer with Al Kαradiation as the excitation source. Binding energies for the high-resolution spectra were calibrated by setting C 1s to 284.6 eV. The UV-Vis absorption spectra and diffuse reflectance spectra were collected on a UV-3600 Shimadzu spectroscope.Photoluminescence spectra (PL) of the samples were obtained at room temperature excited by incident light of 380 nm using a fluorescence spectrometer (Shimadzu RF-5301PC). Electron paramagnetic resonance (EPR) measurements were operated at room temperature, using a Bruker EMX-8/2.7 spectrometer.Elemental analysis (C, H, N) was taken on VARIO EL CUBE microanalyzer.
2.3 Electrochemical Analysis
Electrochemical measurements were performed on a CHI 760 electrochemical workstation in a standard three-electrode cell, using a Pt plate and an Ag/AgCl electrode as counter electrode and reference electrode, respectively41. The working electrodes were prepared by electrophoretic deposition. Two parallel FTO glasses were placed in acetone solution (40 mL)containing 20 mg sample powder and 40 mg iodine, and kept for 5 min with a 10 V bias under potentiostat control. All the samples were deposited on FTO conductive glass with a fixed area of ca 1 cm2, followed by annealing at 150 °C for 1 h. The supporting electrolyte was 50 mL of 0.2 mol·L−1Na2SO4aqueous solution. The transient photocurrent densities (Iph) of the as-prepared electrodes were tested at 0.4 V (vs Ag/AgCl)electrode under visible light irradiation. The visible light generated by a 300 W Xe lamp with a 420 nm cut-off filter chopped manually during the photocurrent analysis. The Nyquist plots of these electrodes were collected by setting the bias voltage at −0.4 V (vs Ag/AgCl) electrode in dark.
2.4 Photocatalytic H2evolution
The photocatalytic H2evolution experiments were carried out in a quartz reaction vessel, by suspending 0.1 g photocatalyst in an aqueous solution (100 mL) containing triethanolamine (10% (volume fraction, φ)). 3% (mass fraction,w) Pt was loaded on the catalystʹs surface by an in-situ photodeposition method using H2PtCl6as the precursor38. A 300 W xenon lamp with a 420 nm cut-off filter generated the visible light. The reactant solution was degassed under a N2flow for 15 min, prior to the irradiation experiment. A flow of cooling water was applied during the photocatalytic reaction to keep the temperature of reaction solution at 20 °C. Gas chromatography (GC) equipped with a thermal conductive detector (TCD) was used to analyze the evolved gas every 1 h.In the recycling experiment, the system was evacuated every 4 h, and this process was repeated for 4 times. The apparent quantum yield (AQY) for H2evolution was measured using 420, 450, 500, 550, 600 and 650 nm band-pass filters. The intensity of irradiation was determined to be ca 0.050 W and the irradiation area was 3.14 cm2. The average intensity of irradiation was measured by Coherent Fieldax-TO spectroradiometer. The AQY was estimated by the following formula:
AQY = (number of evolved H2molecules × 2/number of incident photons) × 100%.
3 Results and discussion
3.1 Structure and composition
Based on Schiff-base chemistry reaction between ―CHO and ―NH2groups, a possible path to incorporate pyridine rings into g-C3N4to form the copolymer, CN-x, is proposed in Fig.S1(Supporting Information). Compared to the pristine g-C3N4, the incorporated pyridine rings are connected by adjacent 3,s-triazine units14.
XRD patterns and FT-IR spectra of CN, CN-x and yGO-CN-3 were recorded to reveal their structure. Both CN-x(Fig.S2(a, b)) (Supporting Information) and yGO-CN-3 (Fig.1a and Fig.S2d) samples show the characteristic peaks of pure g-C3N4, suggesting that the main chemical skeleton of g-C3N4has been well retained. With more pyridine rings being incorporated, the XRD peaks of CN-x become weakened while the FT-IR bands broadened. This phenomenon indicates the chemical environment alteration and periodicity deterioration of the g-C3N4layers/networks to some extent. After combined with GO, yGO-CN-3 composites show intensified signals of g-C3N4in both XRD patterns and FT-IR spectra, as GO may act as a structural promoter via π−π interaction during the direct polymerization of urea. The intensive peak at 10.30°, which corresponds to the (001) reflection of interlayer spacing of GO,is not observable in the composites because of the reduction of GO (Fig.S2c)24.
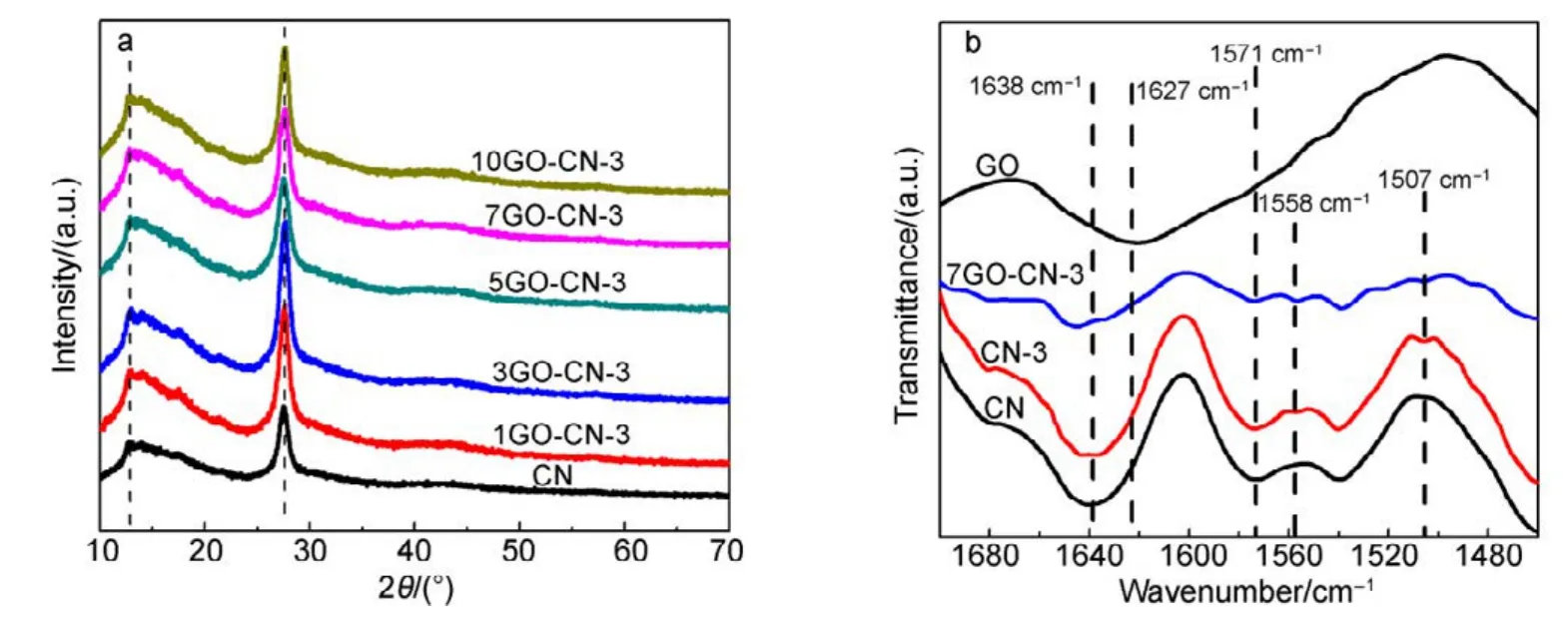
Fig.1 XRD patterns (a) and high-resolution FT-IR spectra (b) of CN, CN-3 and yGO-CN-3 samples.
XRD patterns and FT-IR spectra of CN, CN-x and yGO-CN-3 were recorded to reveal their structure. Both CN-x (Fig.S2(a, b)) (Supporting Information) and yGO-CN-3(Fig.1a and Fig.S2d) samples show the characteristic peaks of pure g-C3N4, suggesting that the main chemical skeleton of g-C3N4has been well retained. With more pyridine rings being incorporated, the XRD peaks of CN-x become weakened while the FT-IR bands broadened. This phenomenon indicates the chemical environment alteration and periodicity deterioration of the g-C3N4layers/networks to some extent. After combined with GO, yGO-CN-3 composites show intensified signals of g-C3N4in both XRD patterns and FT-IR spectra, as GO may act as a structural promoter via π−π interaction during the direct polymerization of urea. The intensive peak at 10.30°, which corresponds to the(001) reflection of interlayer spacing of GO, is not observable in the composites because of the reduction of GO(Fig.S2c)24.
As can be seen from Fig.1b, the high-resolution FT-IR spectra of both CN-3 and 7GO-CN-3 exhibit a distinguishable band located at 1507 cm−1, in accordance with aromatic C=C groups32. Considering the low adding amount of 2,6-pyridinedicarboxaldehyde, it could be preliminarily concluded that pyridine rings have been successfully incorporated into g-C3N4networks. The distinctive peaks of GO at 1732 and 1627 cm−1(Fig.S2d) are ascribed to the C=O stretching mode of COOH and deformation vibration of intercalated H2O, respectively33. It is worth noting that, in the composite yGO-CN-3, these peaks referring to COOH have disappeared while a new peak at 1558 cm−1, which is attributed to the skeletal vibration of graphene sheets, emerges, indicating the formation and presence of rGO in the composite34. The ammonia reductive atmosphere formed during the copolymerization of urea and 2,6-Pyridinedicarboxaldehyde has successfully reduced GO35.
The elemental analysis results of all samples are shown in Table S1 (Supporting Information). The C/N atomic ratio and H content of the polymer increase with more pyridine rings incorporated, which agrees well with our expectation that pyridine can be successfully incorporated into g-C3N4networks to enrich C and H.
TEM was used to unveil the morphologies of CN, CN-3 and 7GO-CN-3 (Fig.2). All samples exhibit typical plateletlike morphology. As for CN and CN-3, curved and distorted layer structure dominates. Due to the low content of rGO, it is hard to find or distinguish graphene layers.

Fig.2 Typical TEM images of (a) CN, (b) CN-3 and (c) 7GO-CN-3.
The surface chemical composition and chemical state of the as-synthesized CN, CN-3, and 7GO-CN-3 were analyzed by XPS. As shown in Fig.3, the three typical signals of C, N,O in CN can be found in all samples. The high resolution C 1s spectra can be deconvoluted into three bands at 284.6, 286.3 and 288.0 eV, which belong to sp2C―C bonds, C―N bonds in carbon species and sp2bonded carbon (N―C=N) in N-containing aromatic rings, respectively. Notably, the sp2C―C band has greatly intensified in 7GO-CN-3, indicating the successful incorporation of rGO in the composite. For the N 1s spectra of these samples, the dominant band at 398.6 eV is attributed to sp2N (C=N―C) in triazine rings,while that at 399.9 eV originates from the tertiary N bonded to carbon atoms in the form of N―(C)3. The band at 401.2 eV is ascribed to amino groups (C―N―H). The other peak centered at 404.2 eV corresponds to the charging effect or positive charge localization in heterocycles. There is little difference in the N 1s spectra of CN-3 and CN, but for 7GO-CN-3, the bands at 399.9 and 401.2 eV are greatly weakened, suggesting the largely decreased amount of N―(C)3and (C―N―H) groups, i.e., the improved polymerization degree of g-C3N4networks36, which favors the photocatalytic H2evolution performance37. Meanwhile,two additional peaks at 400.6 eV and 401 eV emerge in sample 7GO-CN-3, corresponding to the pyrrolic-N and graphitic-N, respect ively38,39. And the pyrrolic-N takes larger percentage than the graphitic-N. Combining the XPS and FT-IR results, we conclude that rGO has been introduced into the g-C3N4matrix and N has been synchronously doped into the rGO networks.
3.2 Photocatalytic activity
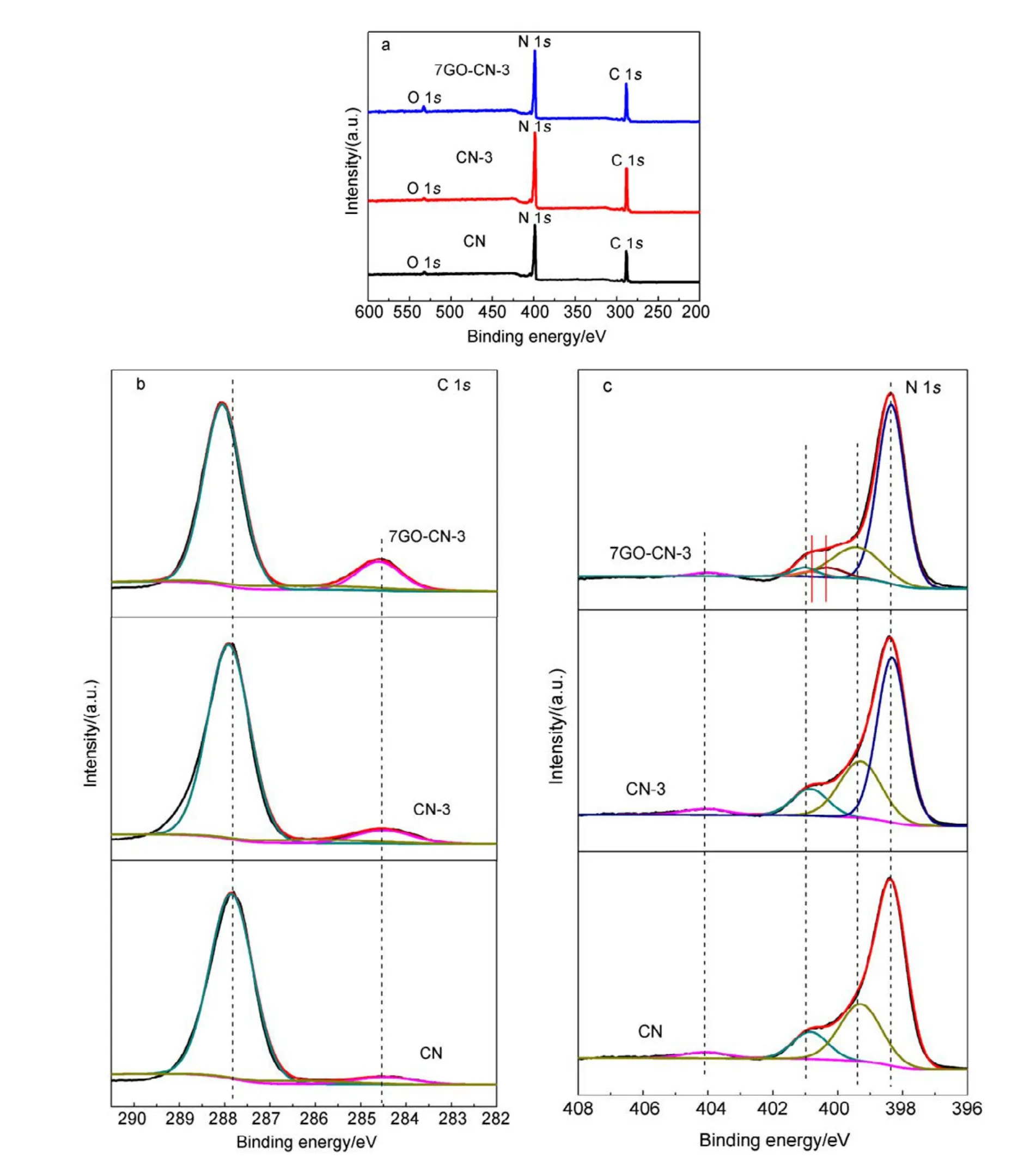
Fig.3 XPS spectra of CN, CN-3 and 7GO-CN-3: (a) survey, (b) C 1s spectra, (c) N 1s spectra.
The photoreduction activity of the obtained samples in water splitting to produce H2was examined under visible light irradiation (λ > 420 nm), using 3% (w) Pt and 10% (φ)triethanolamine as co-catalyst and hole sacrificial agent,respectively. As can be seen in Fig.4a, incorporating an optimized amount of pyridine rings into g-C3N4networks has greatly enhanced the H2evolution activity. CN-3 shows the highest H2evolution rate (98 μmol·h−1), which is about 3.8 times that of pure CN obtained from urea (Fig.4a). Compared with our previous work, quinolone and pyridine modified g-C3N4catalysts exhibit equivalent effects in enhancing the H2evolution activity of g-C3N414.
We chose the recipe of sample CN-3 to further hybridize with GO. Among all the obtained samples, 7GO-CN-3 shows the best photoreduction performance with a H2evolution rate of 304 μmol·h−1(Fig.4b), higher than three times that of CN-3. For both CN-3 and 7GO-CN-3, H2has been produced steadily under the irradiation and there is no significant deactivation during 4 consecutive runs (16 h). The apparent quantum yield (AQY) for H2evolution on 7GO-CN-3 at λ =420, 450, 500, 550, 600, 650 nm were caculated to be 9.6%,6.0%, 3.2%, 3.0%, 2.3% and 1.6%, respectively.Comparatively, the AQY of pure g-C3N4is much lower(1.3% at 420 nm)40.
It is generally known that the surface area of photocatalyst influences much on its activity. It can be seen in Table S2(Supporting Information), the surface area of CN-x does not show much difference from that of CN. After introducing GO, the surface area of the catalysts increases, and 7GOCN-3 has the largest surface area up to 107 m2·g−1, meaning the highest density of accessible catalytic active sites of the sample.
On the other hand, the diffuse reflectance spectra (DRS)(Fig.5a) demonstrate that all CN-x samples possess improved light harvesting capability above 450 nm compared to pure CN. Their band gaps (calculated by Fig.S3(b, c) and listed in Table S2) (Supporting Information) become gradually narrowed with the increasing amount of pyridine rings doped. After the introduction of N-rGO into CN-3, all yGO-CN-3 composite samples exhibit further enhanced visible-light harvesting capability especially in long wavelength region (Fig.5c), though their absorption edges change little. Therefore, 7GO-CN-3 shows considerable AQY at λ = 600 nm and even 650 nm.
By valence band X-ray photoelectron spectroscopy, the intrinsic HOMO energy levels of CN, CN-3 and 7GO-CN-3 were estimated to be 2.46, 2.39 and 2.38 eV, respectively(Fig.S3). Based on their band gaps obtained above, the relative positions of HOMO and LUMO energy levels of CN,CN-3 and 7GO-CN-3 can be calculated. As shown in Fig.S3,the LUMO energy levels of 7GO-CN-3 and CN-3 are 0.04 eV and 0.06 eV higher than those of CN, respectively,resulting in enhanced photoreduction capability of the photo-induced electrons located at them.
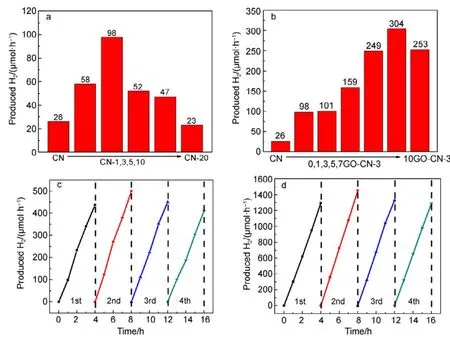
Fig.4 Photocatalytic H2 evolutions on (a) CN and CN-x samples and (b) yGO-CN-3 composites under visible light irradiation(λ > 420 nm); stability tests of H2 evolution on (c) CN-3 and (d) 7GO-CN-3.
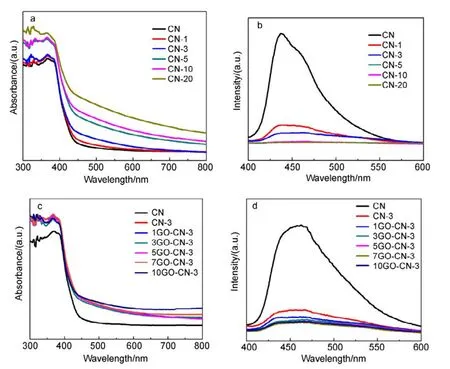
Fig.5 (a, c) UV-Vis absorption spectra and (b, d) PL spectra (excitation wavelength = 380 nm) of all the prepared samples.
To further understand the photophysical behaviors and photoexcited charge carriers of CN and all the modified samples, their photoluminescence (PL) spectra were recorded(Fig.5(b, d)). They all exhibit two distinguished PL bands/shoulder bands41. The one at around 440 nm is ascribed to the intrinsic LUMO to HOMO emission of g-C3N4, corresponding to the samplesʹ slightly different band gaps. The other weak shoulder band at around 463 nm results from charge transfer emission. The intensity of PL bands shows a significant decrease at increased amount of pyridine rings incorporated,and decreases further after N-rGO incorporation. Generally,this phenomenon suggests the decreased luminous recombination probability of the photo-induced charge carriers in the composite photocatalysts, certainly favorable for charge separation and reactivity enhancement in photocatalytic reactions42.
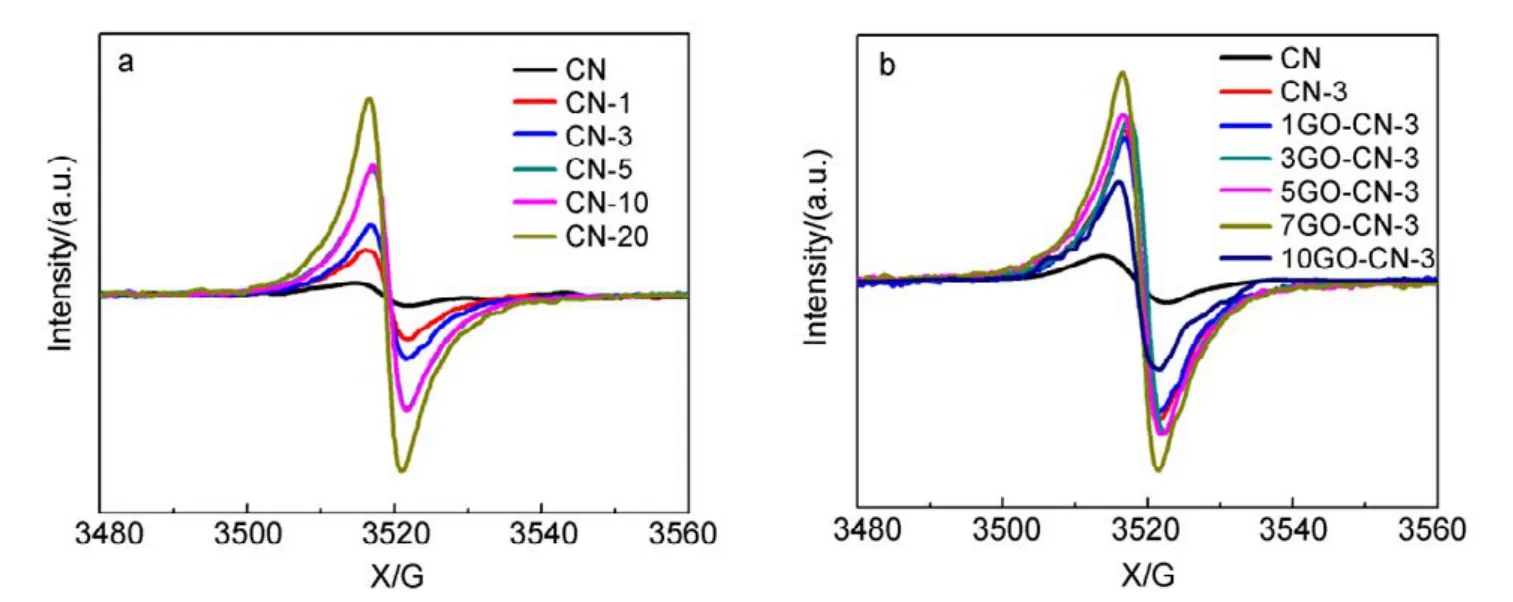
Fig.6 Room temperature EPR spectra of CN, CN-x and yGO-CN-3.
The X-band electron paramagnetic resonance (EPR)analysis was performed to further explore the electronic structure of CN, CN-x and yGO-CN-3. It can be seen in Fig.6 that all samples exhibit a symmetric signal with a g value centered at around 2.0040, indicating the existence of unpaired electrons on nitrogen atoms. For g-C3N4, the unpaired electrons on nitrogen atoms can be stabilized to a large extent through p−π conjugation with 3,s-triazine ring.With more pyridine rings being incorporated, the EPR signals gradually increase, indicating the broadened delocalization region of the electrons in the copolymers, thus the radicals will acquire enhanced stability. All the yGO-CN-3 samples show intensified EPR signals in comparison with CN, among which 7GO-CN-3 has the highest signal intensity. This signal enhancement indicates the increased amount of photochemical radical pairs, the broadened delocalization of the electrons and the significantly improved stability of radicals in the incorporated samples. Photoelectrochemical tests were performed to investigate the separation of photogenerated electrons and holes, and the interfacial charge transfer in the photocatalysts. A gradual but marked radius decrease of the semicircular Nyquist plots (Fig.7a) is observed for CN-3 and 7GO-CN-3 in dark, indicating its accelerated interface charge transport43. The photocurrent Iph(Fig.7b) of 7GO-CN-3 shows significant enhancements over those of CN-3 and CN. The Iphof 7GO-CN-3 (1.1 μA·cm−2) is about twice and 4 times that of CN-3 (0.6 μA·cm−2) and CN (0.3 μA·cm−2) under visible light irradiation, indicating the much more effective charge separation and therefore the higher photocatalytic activity of 7GO-CN-3 than those of CN-3 and CN44.
3.3 Photocatalytic mechanism
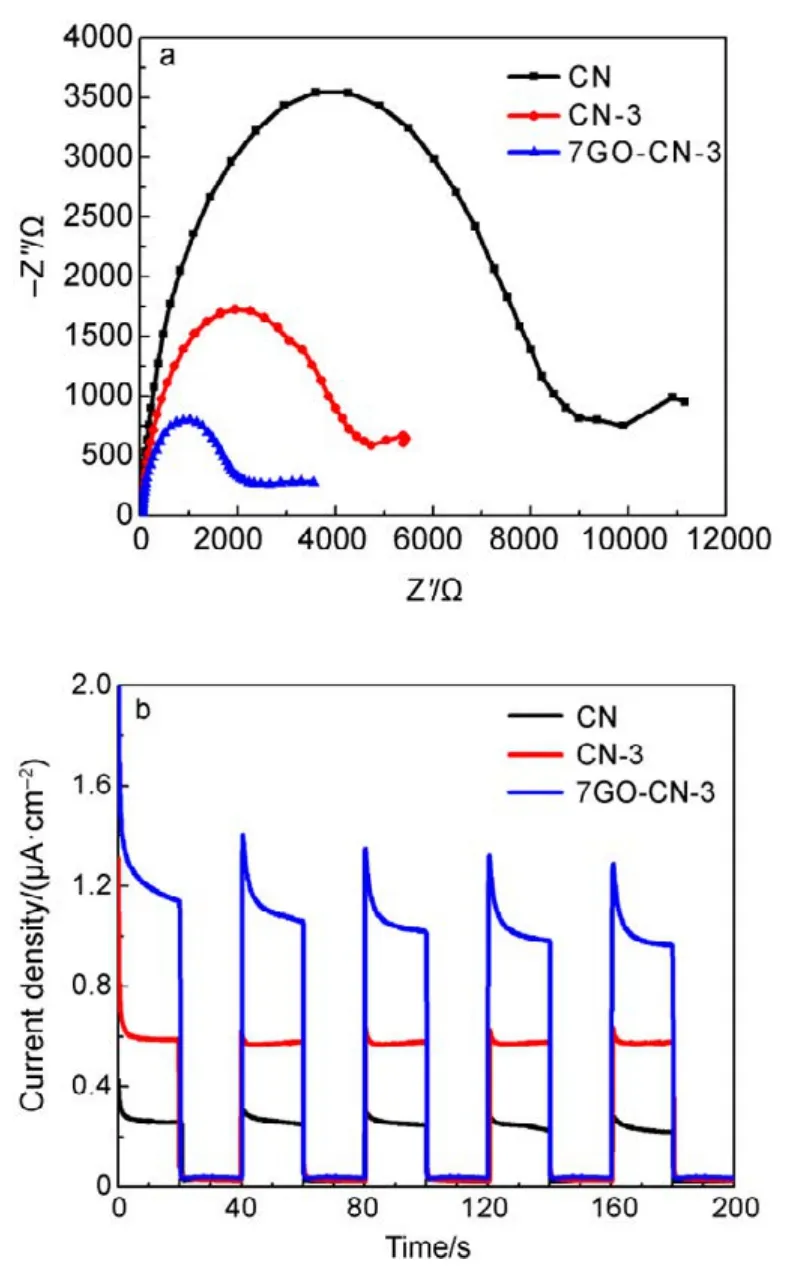
Fig.7 Photoelectrochemical properties of CN, CN-3 and 7GO-CN-3: (a) electrochemical impedance spectroscopy (EIS)Nyquist plots in dark and (b) periodic on/off photocurrent responses under visible-light irradiation.
Generally, in g-C3N4loaded with Pt cocatalyst, electrons are excited from the intrinsic HOMO (valence band (VB)) to LUMO (conduction band (CB)) by incident photons. The photogenerated electrons on the CB of g-C3N4transfer to Pt nanoparticles, where the activated H2O can be reduced to H2,while the holes on the VB will migrate to oxidize TEA to TEA+. On the contrast, in the donor-acceptor conjugated g-C3N4copolymer (pyridine modified g-C3N4in this case),nitrogen groups, which have lone pair electrons, act as electron donor, while aromatic rings take the role of electron acceptor45. Specifically, the photoexcited electrons will transfer from the donor across the intramolecular interface to the acceptor, resulting in improved separation and prolonged lifetime of charge carriers46. Meanwhile, the elevated LUMO energy level of pyridine-modified g-C3N4makes the photoinduced electrons more reductive (Fig.S3).
It is believed that rGO works as not only a sensitizer to enhance the light absorbance, but also a “fast channel” for the rapid transfer of photogenerated electrons27. More significantly, the introduction of neighbor N dopants could change the spin density and charge distribution of carbon atoms, forming “electron-mobility activation region” on graphene surface for the rapid and effective transfer of photogenerated electrons47. And the pyrrolic N atoms at the edges or on the defects of rGO offer strong attraction and high mobility for photogenerated electrons48.
Based on the above analysis, a synergetic photocatalytic mechanism of pyridine-modified g-C3N4/N-rGO composites is proposed and shown in Fig.8. Under visible light irradiation, electron and hole pairs are generated in this composite. The photoexcited electrons generated in pyridine-modified g-C3N4tend to transfer to the pyridine ring acceptor or diffuse across the interface between pyridine-modified g-C3N4and rGO to the graphitic/pyrrolic nitrogen atoms doped in rGO. Then the electrons transfer further to Pt NPs located at the surface of pyridine-modified g-C3N4or rGO to reduce H2O to H2. Meanwhile, the holes remaining in pyridine-modified g-C3N4will oxidize TEA to TEA+. This composite system with pyridine rings as intramolecular electron acceptor, N-doped rGO as “electrontransfer activation region”, and Pt as electron traps demonstrates greatly promoted charge separation, transfer and surface photocatalytic reactivity. As a consequence, the photocatalytic activity of this pyridine-modified g-C3N4/N-rGO composite is remarkably enhanced.
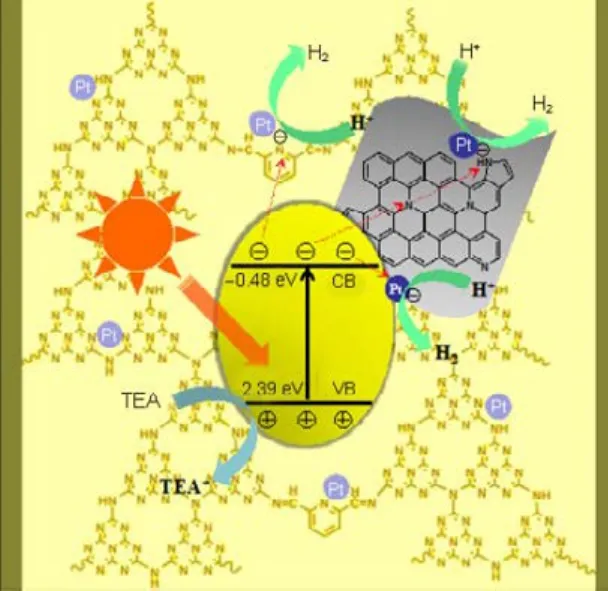
Fig.8 Schematic diagrams of the proposed synergetic photocatalytic mechanism of the pyridine modified g-C3N4/N-rGO composite.
4 Conclusions
In summary, we have successfully constructed a photocatalyst by incorporating N-doped reduced graphene oxide (N-rGO) into pyridine-modified g-C3N4via a simple one-pot calcination route. Pyridine-copolymerization results in the significant structural and electronic modification of g-C3N4, including promoted electron transfer from the intramolecular donor to acceptor, elevated conduction band level (enhanced reducibility of photogenerated electrons) and improved separation ability of charge carriers. The incorporation of N-rGO remarkably enhances the visible light absorbance, enlarges the surface area and creates an“electron-mobility activation region” for fast electron transfer. The peak H2production rate on this composite reaches 304 μmol·h−1, about 11.7 and 3.1 times of those on pure g-C3N4obtained from urea and pyridine-modified g-C3N4, respectively. This work not only provides a costeffective and facile strategy to fabricate g-C3N4or graphene based non-mental catalysts, but also a promising photocatalyst candidate for future solar fuel production. And this composite system integrating intramolecular electron acceptor with intermolecular fast channel for electron transfer provides an effective strategy for the rational design and construction of highly active photocatalysts.
Supporting Information: available free of charge via the internet at http://www.whxb.pku.edu.cn.
(1) Fujishima, A.; Honda, K. Nature 1972, 238, 474.doi: 10.1038/238037a0
(2) Zhang, G.; Kim, G.; Choi, W. Energy Environ. Sci. 2014, 7, 954.doi: 10.1039/c3ee43147a
(3) Ran, J. R.; Zhang, J.; Yu, J. G.; Jaroniec, M.; Qiao, S. Z. Chem. Soc.Rev. 2014, 43, 7787. doi: 10.1039/c3cs60425j
(5) Schwinghammer, K.; Tuffy, B.; Mesch, M. B.; Wirnhier, E.;Martineau, C.; Taulelle, F.; Schnick, W.; Senker, J.; Lotsch, B. V.Angew. Chem. Int. Ed. 2013, 52, 2435. doi: 10.1002/anie.201206817
(6) Wang, X. C.; Maeda, K.; Thomas, A.; Takanabe, K.; Xin, G.;Carlsson, J. M.; Domen, K.; Antonietti, M. Nat. Mater. 2009, 8, 76.doi: 10.1038/NMAT2317
(7) Zhang, G. G.; Lan, Z. A.; Wang, X. C. Angew. Chem. Int. Ed. 2016,55, 15712. doi: 10.1002/anie.201607375
(8) Zhang, G. G.; Lan, Z. A.; Lin, L. H.; Lin, S.; Wang, X. C. Chem. Sci.2016, 7, 3062. doi: 10.1039/c5sc04572j
(9) Mamba, G.; Mishra, A. K. Appl. Catal. B: Environ. 2016, 198, 347.doi: 10.1016/j.apcatb.2016.05.052
(10) Wang, X. C.; Chen, X. F.; Thomas, A.; Fu, X. Z.; Antonietti, M. Adv.Mater. 2009, 21, 1609. doi: 10.1002/adma.200802627
(11) Hu, S. Z.; Chen, X.; Li, Q.; Li, F. Y.; Fan, Z. P.; Wang, H.; Wang, Y.J.; Zheng, B. H.; Wu, G. Appl. Catal. B: Environ. 2017, 201, 58.doi: 10.1016/j.apcatb.2016.08.002
(12) Ho, W. K.; Zhang, Z. Z.; Lin, W.; Huang, S. P.; Zhang, X. W.; Wang,X. X.; Huang, Y. ACS. Appl. Mater. Interfaces 2015, 7, 5497.doi: 10.1021/am509213x
(13) Zhang, J. S.; Chen, X. F.; Takanabe, K.; Maeda, K.; Domen, K.;Epping, J. D.; Fu, X. Z.; Antonietti, M.; Wang, X. C. Angew. Chem.Int. Ed. 2010, 49, 441. doi: 10.1002/anie.200903886
(14) Fan, X. Q.; Zhang, L. X.; Wang, M.; Huang, W. M.; Zhou, Y. J.; Li,M. L.; Cheng, R. L.; Shi, J. L. Appl. Catal. B: Environ. 2016, 182, 68.doi: 10.1016/j.apcatb.2015.09.006
(15) Tian, J. J.; Zhang, L. X.; Fan, X. Q.; Zhou, Y. J.; Wang, M.; Cheng,R. L.; Li, M. L.; Kan, X. T.; Jin, X. X.; Liu, Z. H.; Gao, Y. F.; Shi, J.L. J. Mater. Chem. A 2016, 4, 13814. doi: 10.1039/c6ta04297j
(16) Zhang, J. S.; Zhang, M. W.; Sun, R. Q.; Wang, X. C. Angew. Chem.Int. Ed. 2012, 51, 10145. doi: 10.1002/anie.201205333
(17) Hou, Y. D.; Laursen, A. B; Zhang, J. S; Zhang, G. G; Zhu, Y. S;Wang, X. C.; Dahl, S.; Chorkendorff, I. Angew. Chem. Int. Ed. 2013,52, 3621. doi: 10.1002/anie.201210294
(18) Sun, L. M.; Qi, Y.; Jia, C. J.; Jin, Z.; Fan, W. L. Nanoscale 2014, 6,2649. doi: 10.1039/c3nr06104c
(19) Geim, A. Science 2009, 324, 1530. doi: 10.1126/science.1158877
(20) Hou, Y.; Wen, Z. H.; Cui, S. M.; Guo, X. R.; Chen, J. H. Adv. Mater.2013, 25, 6291. doi: 10.1002/adma.201303116
(21) Huang, Q. W.; Tian, S. Q.; Zeng, D. W.; Wang, X. X.; Song, W. L.;Li, Y. Y.; Xiao, W.; Xie, C. S. ACS Catal. 2013, 3, 1477.doi: 10.1021/cs400080w
(22) Zhang, F. W.; Wen, Q. J.; Hong, M. Z.; Zhuang, Z. Y.; Yu, Y. Chem.Eng. J. 2017, 307, 593. doi: 10.1016/j.cej.2016.08.120
(23) Li, X.; Yu, J. G.; Low, J. X.; Fang, Y. P.; Xiao, J.; Chen, X. B.J. Mater. Chem. A 2015, 3, 2485. doi: 10.1039/c4ta04461d
(24) Liao, G. Z.; Chen, S.; Quan, X.; Yu, H. T.; Zhao, H. M. J. Mater.Chem. 2012, 22, 2721. doi: 10.1039/c1jm13490f
(25) Wang, P.; Wang, J.; Wang, X. F.; Yu, H. G.; Yu, J. G.; Lei, M.;Wang, Y. G. Appl. Catal. B: Environ. 2013, 132-133, 452.doi: 10.1016/j.apcatb.2012.12.009
(26) Xiang, Q. J.; Yu, J. G.; Jaroniec, M. J. Phys. Chem. C 2011, 115,7355. doi: 10.1021/jp200953k
(27) Ong, W. J.; Tan, L. L.; Chai, S. P.; Yong, S. T. Chem. Commun. 2015,51, 858. doi: 10.1039/c4cc08996k
(28) Li, Y. B.; Zhang, H. M.; Liu, P. R.; Wang, D.; Li, Y.; Zhao, H. J.Small 2013, 9, 3336. doi: 10.1002/smll.201203135
(29) Liu, Q.; Zhang, J. Y. Langmuir 2013, 29, 3821.doi: 10.1021/la400003h
(30) Gusain, R.; Kumar, P.; Sharma, O. P.; Jain, S. L.; Khatri, O. P. Appl.Catal. B: Environ. 2016, 181, 352. doi: 10.1016/j.apcatb.2015.08.012
(31) Zheng, Y.; Jiao, Y.; Zhu, Y. H.; Li, L. H.; Han, Y.; Chen, Y.; Du, A.J.; Jaroniec, M.; Qiao, S. Z. Nat. Commun. 2014, 5, 3783.doi: 10.1038/ncomms4783
(32) Fan, X. Q.; Zhang, L. X.; Cheng, R. L.; Wang, M.; Li, M. L.; Zhou,Y. J.; Shi, J. L. ACS Catal. 2015, 5, 5008.doi: 10.1021/acscatal.5b01155
(33) Guo, H. L.; Wang, X. F.; Qian, Q. Y.; Wang, F. B.; Xia, X. H. ACS Nano 2009, 3, 2653. doi: 10.1021/nn900227d
(34) Xiang, Q. J.; Yu, J. G.; Jaroniec, M. J. Phys. Chem. C 2011, 115,7355. doi: 10.1021/jp200953k
(35) Yuan, B.; Wei, J. X.; Hu, T. J.; Yao, H. B.; Jiang, Z. H.; Fang, Z. W.;Chu, Z. Y. Chin. J. Catal. 2015, 36, 1009.doi: 10.1016/S1872-2067(15)60844-0
(36) Cui, Y.J.; Zhang, G. G.; Lin, Z. Z.; Wang, X. C. Appl. Catal. B:Environ. 2016, 181, 413. doi: 10.1016/j.apcatb.2015.08.018
(37) Martin, D. J.; Qiu, K. P.; Shevlin, S. A.; Handoko, A. D.; Chen, X.W.; Guo, Z. X.; Tang, J. W. Angew. Chem. Int. Ed. 2014, 53, 9240.doi: 10.1002/anie.201403375
(38) Wang, D. W.; Su, D. S. Energy Environ. Sci. 2014, 7, 576.doi: 10.1039/c3ee43463j
(39) Zhou, Y. J.; Zhang, L. X.; Huang, W. M.; Kong, Q. L.; Fan, X. Q.;Wang, M.; Shi, J. L. Carbon 2016, 99, 111.doi: 10.1016/j.carbon.2015.12.008
(40) Zhang, X. H.; Yu, L. J.; Zhuang, C. S.; Peng, T. Y.; Li, R. J.; Li, X. G.ACS Catal. 2014, 4, 162. doi: 10.1021/cs400863c
(41) Bledowski, M.; Wang, L. D.; Ramakrishnan, A.; Khavryuchenko, O.V.; Khavryuchenko, V. D.; Ricci, P. C.; Strunk, J.; Cremer, T.;Kolbeck, C.; Beranek, R. Phys. Chem. Chem. Phys. 2011, 13, 21511.doi: 10.1039/c1cp22861g
(42) Li, M. L.; Zhang, L. X.; Fan, X. Q.; Zhou, Y. J.; Wu, M. Y.; Shi, J. L.J. Mater. Chem. A 2015, 3, 5189. doi: 10.1039/c4ta06295g
(43) Liang, Q. H.; Li, Z.; Yu, X. L.; Huang, Z. H.; Kang, F. Y.; Yang, Q.H. Adv. Mater. 2015, 27, 4634. doi: 10.1002/adma.201502057
(44) Li, M. L.; Zhang, L. X.; Wu, M. Y.; Du, Y. Y.; Fan, X. Q.; Wang, M.;Zhang, L. L.; Kong, Q. L.; Shi, J. L. Nano Energy 2016, 19, 145.doi: 10.1016/j.nanoen.2015.11.010
(45) Grabowski, Z.; Rotkiewicz, K. Chem. Rev. 2003, 103, 3899.doi: 10.1021/cr940745l
(46) Li, M. L.; Zhang, L. X.; Fan, X. Q.; Wu, M. Y.; Du, Y. Y.; Wang, M.;Kong, Q. L.; Zhang, L. L.; Shi, J. L. Appl. Catal. B: Environ. 2016,190, 36. doi: 10.1016/j.apcatb.2016.02.060
(47) Xu, Y.; Mo, Y. P.; Tian, J.; Wang, P.; Yu, H. G.; Yu, J. G. Appl.Catal. B: Environ. 2016, 181, 810. doi: 10.1016/j.apcatb.2015.08.049
(48) Strelko, V. V.; Kuts, V. S.; Thrower, P. A. Carbon 2000, 38, 1499.doi: 10.1016/S0008-6223(00)00121-4
Incorporation of N-Doped Reduced Graphene Oxide into Pyridine-Copolymerized g-C3N4for Greatly Enhanced H2Photocatalytic Evolution
CHENG Ruo-Lin1,2JIN Xi-Xiong1,2FAN Xiang-Qian1,2WANG Min1,2TIAN Jian-Jian1,2ZHANG Ling-Xia1,2,*SHI Jian-Lin1,2,*
(1State Key Laboratory of High Performance Ceramics and Superfine Microstruture, Shanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai 200050, P. R. China;2University of Chinese Academy of Sciences, Beijing 100049, P. R. China)
Here, we fabricated a pyridine-copolymerized g-C3N4by a novel and cost-effective approach based on Schiff-base chemistry. Thus produced g-C3N4showed significantly enhanced and stable visible-light photocatalytic H2evolution performance compared to pristine g-C3N4obtained from urea.Subsequently, we constructed a composite of pyridine-modified g-C3N4and N-doped reduced graphene oxide (N-rGO) by facile one-pot calcination to elevate the photocatalytic efficiency further. The peak H2production rate achieved using this composite was 304 µmol·h−1, about 11.7 and 3.1 times as those obtained using pure g-C3N4and pyridine-modified g-C3N4, respectively. In addition to enhanced visible light absorbance and enlarged surface area, the promoted separation, transfer, and surface reactivity of photogenerated charge carriers by the pyridine ring as intramolecular electron acceptor and N-rGO as“electron-transfer activation region” are considered responsible for the remarkably enhanced photocatalytic activity.
Photocatalysis; Graphitic carbon nitride; Copolymerization; Hydrogen evolution;Nanocomposite
December 27, 2016; Revised: March 15, 2017; Published online: April 7, 2017.
O649
10.1038/35104607
[Article]
10.3866/PKU.WHXB201704076 www.whxb.pku.edu.cn
*Corresponding authors. SHI Jian-Lin, Email: jlshi@mail.sic.ac.cn; Tel: +86-21-52412706. ZHANG Ling-Xia, Email: zhlingxia@mail.sic.ac.cn;Tel: +86-21-52412706.
The project was supported by the State Key Program for Basic Research of China (2013CB933200).
国家重点基础研究发展规划项目(2013CB933200)资助
© Editorial office of Acta Physico-Chimica Sinica
猜你喜欢
安徽化工(2022年1期)2022-02-15
中华养生保健(2020年3期)2020-11-16
化工学报(2020年4期)2020-05-28
化工设计(2020年2期)2020-05-01
陶瓷学报(2019年6期)2019-10-27
今日农业(2019年11期)2019-08-13
郑州大学学报(理学版)(2017年1期)2017-04-07
化工管理(2017年25期)2017-03-05
浙江农业科学(2016年11期)2016-05-04
应用化工(2014年11期)2014-08-16
