
环状磷酸酯作为电解液多功能添加剂的量子化学计算研究
2017-10-24 11:28项宏发贺晓东
合肥工业大学学报(自然科学版) 2017年9期
项宏发, 林 敏, 郑 浩, 贺晓东
(合肥工业大学 材料科学与工程学院,安徽 合肥 230009)
环状磷酸酯作为电解液多功能添加剂的量子化学计算研究
项宏发, 林 敏, 郑 浩, 贺晓东
(合肥工业大学 材料科学与工程学院,安徽 合肥 230009)
环状磷酸酯作为锂离子电池电解液多功能添加剂倍受关注,研究其结构与性能的构效关系对于相关电解液的组成设计和应用具有重要意义。文章采用量子化学方法计算了15种环状磷酸酯的前线轨道能量、化学硬度、氧化还原电位等量子化学参数,从理论上预测其用作锂离子电池电解液添加剂的可能性。研究了环状磷酸酯分子结构与电化学稳定性之间的关系。计算结果表明:苯环取代不仅可以提高还原电位,还可以显著降低氧化电位和化学硬度,提高分子的反应活性;氟原子取代可以提高分子的还原电位,对氧化电位和化学硬度影响并不明显。
锂离子电池;电解液;添加剂;磷酸酯;量子化学
锂离子电池由于具有能量密度大、工作电压高、循环寿命长、无环境污染等优点,被广泛用于小型电子设备中,成为近20年来商业化最成功的二次电池[1]。目前,锂离子电池应用于电动汽车的最大阻碍是安全问题,主要由电解液的性能决定。电解液一般由有机溶剂、锂盐、添加剂按一定比例配制而成。其中,电解液添加剂由于用量少、作用大成为电池领域研究热点,也是电解液应用的核心技术。根据功能不同,电解液添加剂一般分为成膜添加剂、抗过充添加剂、阻燃添加剂、电解质稳定添加剂等。为了获得良好的电池综合性能,电解液中往往包含多种添加剂。多种添加剂的引入一方面提高了电池成本,另一方面使电解液成分更为复杂,各组分之间的相互影响导致不能达到预期效果。因此,集多种功能于一身的多功能添加剂具有成本与技术应用优势,相关研究对于电解液开发应用具有重要意义。
以磷酸三甲酯(TMP)[2]为代表的含磷有机化合物具有很好的阻燃效果和氧化稳定性,但是与石墨负极的兼容性较差。文献[3]发现环状结构的乙烯基乙基磷酸酯(EEP)可以提高TMP基阻燃电解液与石墨负极的兼容性,并在石墨负极形成稳定的固体电解质界面膜(SEI)。文献[4]利用理论计算描述环状磷酸酯EEP与非环状磷酸酯TMP明显不同的量子化学特征,通过与环状的碳酸乙烯酯(EC)的比较说明其作为多功能添加剂的可能性,并通过实验证明在基准电解液中添加10% EEP后,LiNi1/3Co1/3Mn1/3O2|Li电池在0.1C的电流下过充至6 V的时间延缓了10 h,首次库伦效率从86.16%提高到89.23%,电解液自熄灭时间减少了50%。可见,环状磷酸酯EEP不但具有阻燃功能,还具有在石墨负极成膜的功能,并且在高电压下可开环聚合实现抗过充的功能,因此作为多功能添加剂具有重要的应用前景。
目前,电解液添加剂的研究往往建立在实验的基础上,通过大量的试探性实验来发掘添加剂的功能,需要通过排列组合设计众多的实验,实验时将耗费大量的人力和物力,而最终具有实用价值或应用潜力的电解液却寥寥无几。而本文在EEP的研究基础上[4],通过氟原子取代、苯环取代、碳碳双键取代等方式构建了14个EEP的同系物,计算包括EEP在内的15个环状磷酸酯和EC的最高占有分子轨道(highest occupied molecular orbital,HOMO)能量、最低未占有分子轨道(lowest unoccupied molecular orbital,LUMO)能量、化学硬度、偶极矩、氧化还原电位、锂离子结合能等参数,通过比较筛选出类似于EEP具有用作多功能添加剂潜力的环状磷酸酯分子。
1 理论背景
在分析计算获得的量子化学参数之前,有必要对部分参数,如绝热电子亲和势、电离能、氧化还原电位和化学硬度等概念作简要介绍。
绝热电子亲合势(EA)是一个中性分子(M)与一个电子结合形成并发生结构弛豫所释放的能量,可以用中性分子(M)和负离子(M-)之间的电子能量差来计算,即
EA=E(M)-E(M-)
(1)
电离能(EI)是从一个中性分子(M)中移出的一个电子形成阳离子(M+)所需要的能量,为阳离子与中性分子的电子能量差,即
EI=E(M+)-E(M)
(2)
锂离子电池电解液成膜添加剂分子通常具有较高的还原电位,得电子能力较强,可以优先于溶剂分子被还原,因而有利于在负极表面形成SEI膜,标准的还原电位(ER)可由中性分子(M)与其负离子(M-)的吉布斯自由能之差计算,为了获得对Li/Li+的相对电极电位,减去1.46 V[5],具体如下:
ER=[G(M)-G(M-)]/e-1.46
(3)
抗过充添加剂一般具有较低的氧化电位,可以优先于碳酸酯溶剂被氧化形成导电聚合物,在电池内部形成电子通路,降低电压,提高电池的抗过充性能。标准的氧化电位E0是由中性分子(M)与其正离子(M+)的吉布斯自由能之差决定的,减去1.46 V即得到对Li/Li+的相对电极电位,即
E0=[G(M+)+G(M)]/e-1.46
(4)
电子能量E与吉布斯自由能G之间的关系为:
G=E+RT-TS
(5)
其中,R为摩尔气体常数;T为热力学温度;S为熵,可以将吉布斯自由能理解为电子能量的热力学修正,更接近于实际情况。化学硬度[6]对应于分子发生化学反应的活泼性。对于理想的负极成膜添加剂或抗过充添加剂,一般具有较低的化学硬度,在较低的电位下,容易在负极表面发生还原反应[7];或在正极表面容易发生氧化反应。化学硬度(η)的定义为:
η=(ELUMO-EHOMO)/2
(6)
为了筛选出具有成膜和抗过充添加剂潜力的分子,结合电解液添加剂量子化学参数与电化学功能的关系,依据文献[5-7]的报道明确多功能电解液添加剂筛选标准如下:① 还原电位高于电解液主要溶剂EC,优先于EC被还原,具有在石墨负极表面优先形成SEI膜的可能性;② 氧化电位低于EC,优先于EC被氧化,发生开环反应在正极表面形成聚合物,具有用作抗过充添加剂的可能性;③ 化学硬度低于EEP,反应活性越高,EEP作为已知的多功能添加剂,比EEP化学硬度更低意味着反应活性更高,具有作为多功能添加剂的潜力。
2 计算细节
本文中所有的计算均由Gaussian 09软件完成,计算的理论方法均采用B3PW91,一种常用的密度泛函理论方法。密度泛函理论具有精度高、计算速度快等特点,在量子化学计算中已得到广泛应用。分子的结构优化和频率计算选用6-31G(d)基组,分子优化完成后的单点能计算采用高等级的6-311++G(d,p) 基组。本文使用不同等级的基组组合可以提高计算的效率,同时保证了计算的精确度。为了模拟出电解液添加剂所处的溶液环境,必须考虑溶剂化效应,采用极化连续模型(polarizable continuum model,PCM)。PCM将溶剂看作连续的极化介质,介质中存在一系列由球体重叠而成的空腔,溶质分子(或离子)则填充在这些空腔中。溶剂化参数中的介电常数参考常用的基准电解液选取46.6,所有的计算均在该模型和参数下完成。
3 结果与讨论
14种EEP的同系物与EEP、EC分子的结构简式如图1所示,图1中,1~14分别为乙烯基甲基磷酸酯、乙烯基乙基亚磷酸酯、丁烯基乙基磷酸酯、1,2-丁烯基乙基磷酸酯、乙炔基乙基磷酸酯、乙烯基三氟代甲基磷酸酯、乙烯基1-三氟代乙基磷酸酯、氟代乙烯基乙基磷酸酯、1-三氟代丙烯基乙基磷酸酯、苯基乙基磷酸酯、苯乙烯基乙基磷酸酯、二苯基乙基磷酸酯、丙烯基乙基磷酸酯、甲基乙基二磷酸酯。
这16个分子的HOMO能量、LUMO能量、化学硬度、还原电位、氧化电位等量子化学参数见表1所列。首先,从还原电位来看,分子6~12与14的还原电位在0.07~1.59 V之间,高于EC的还原电位(0.05 V)。除分子14外,分子6~12都含有苯环或氟原子取代基,而还原电位低于EC的分子1~5与13则无苯环或氟原子取代基,都是EEP通过双键或烷基取代得到的衍生物,这意味着在EEP的结构中加入苯环或氟原子取代都可以提高分子的还原电位。其次,从氧化电位来看,所有候选分子的氧化电位在4.49~6.75 V之间,均低于EC的氧化电位6.86 V;但是含氟原子取代基的分子6~9的氧化电位分别为6.75、6.77、6.49、6.51 V,十分接近于EC的6.86 V;而有苯环取代的分子10~12分别为4.89、5.31、4.49 V,都远低于EC的氧化电位6.86 V。最后,从化学硬度来看,低于EEP化学硬度4.10 eV且经过还原电位筛选的分子只有10~12,这3个分子都具有苯环取代基,化学硬度的值分别为3.01、3.15、2.27 eV。经过以上分析,本文筛选出了分子10~12有用作成膜和抗过充电解液添加剂的潜力,发现EEP同系物分子的量子化学参数有明显的官能团特征,苯环取代不仅可以提高还原电位,还可以显著降低氧化电位和化学硬度,提高分子的反应活性;氟原子取代可以提高分子的还原电位,但是对氧化电位和化学硬度影响并不明显。
量子化学前线轨道理论[8-9]认为:分子在反应过程中,优先起作用的是前线轨道,前线轨道在很大程度上反映了分子的物理化学性质。LUMO能量对应于分子的还原电位,LUMO能量越低,还原电位越高。计算目标物质的LUMO能量是寻找优良成膜添加剂的重要方法。HOMO能量对应于分子的氧化电位,HOMO越高,氧化电位越低。
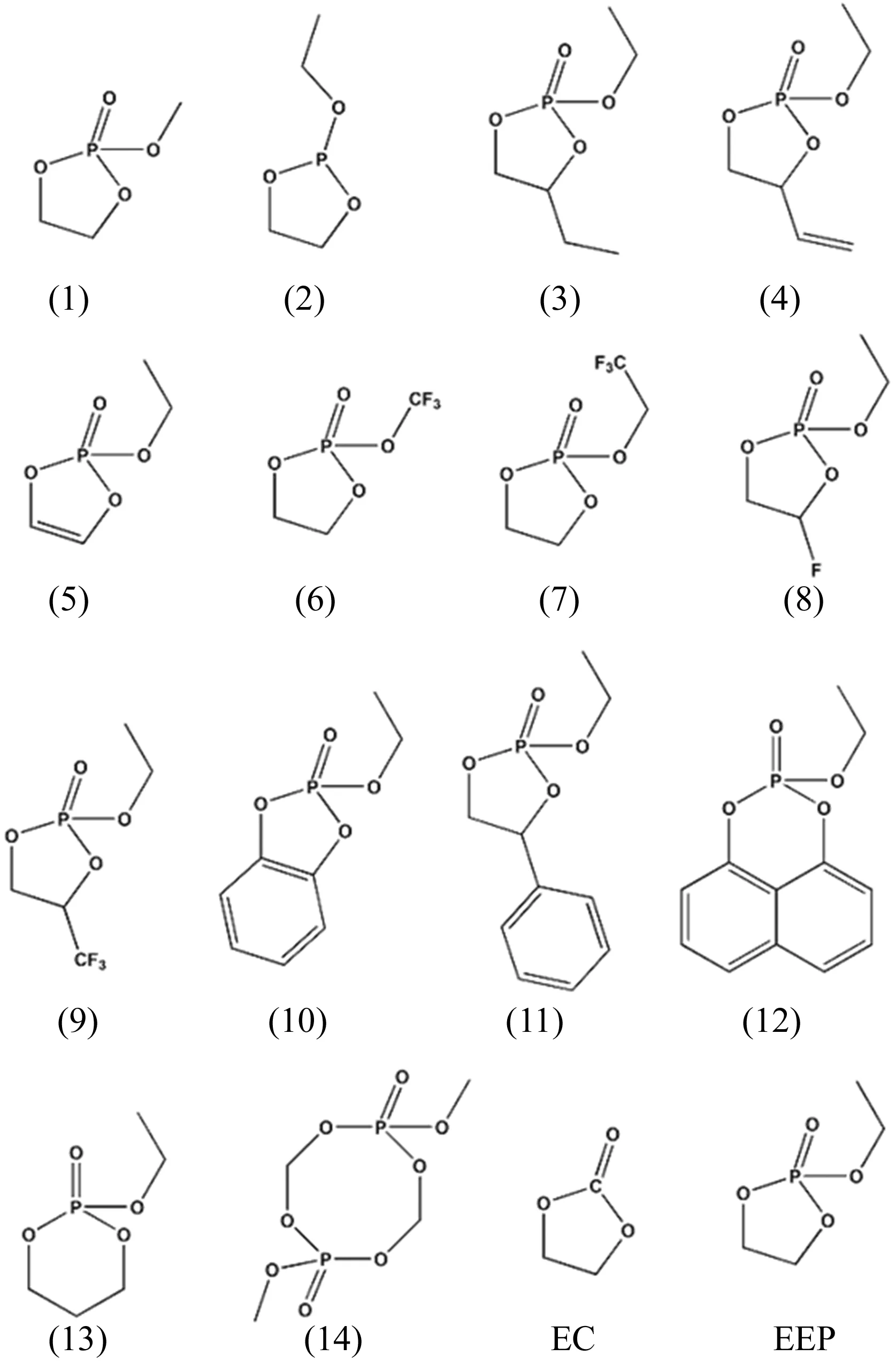
图1 14种EEP分子同系物与EEP、EC的结构简式
计算目标物质的HOMO能量,对于添加剂抗过充性能的考察能够提供有益的参考。根据这16个分子的计算结果,分别对LUMO能量与还原电位、HOMO能量与氧化电位作线性回归分析,HOMO能量与氧化电位E0、LUMO能量与还原电位ER的关系如图2、图3所示。
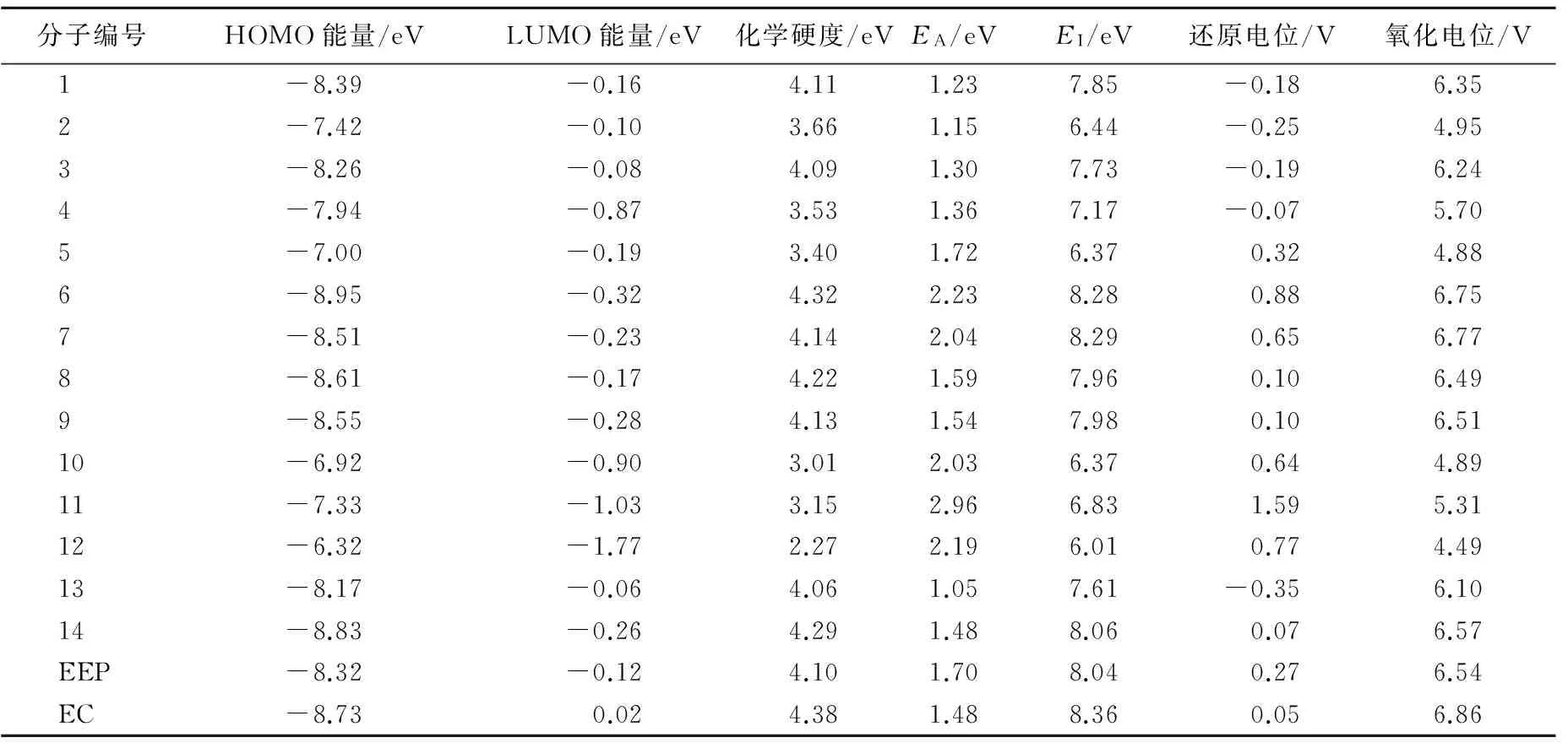
表1 14种EEP分子同系物与EEP、EC的量子化学参数
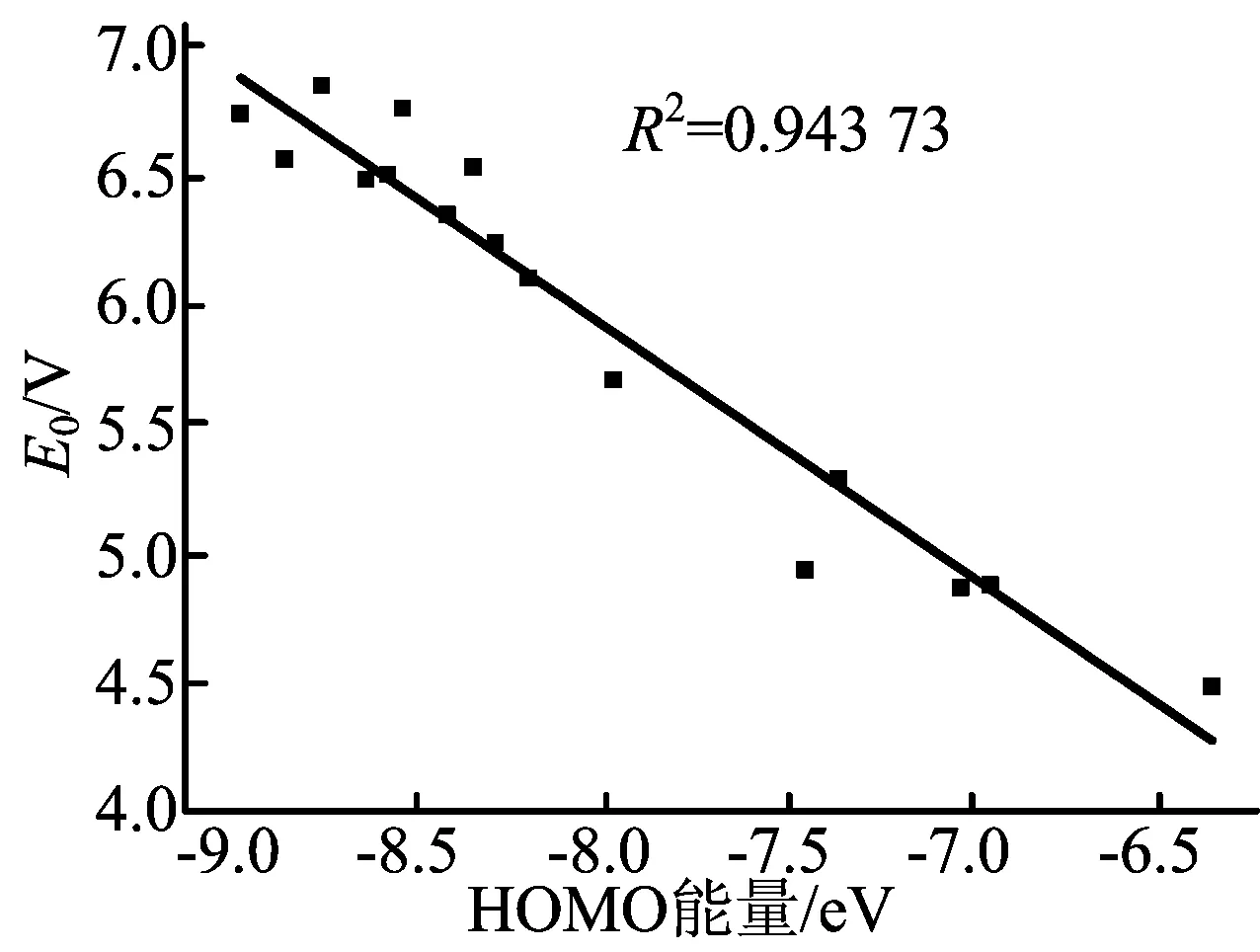
图2 HOMO能量与氧化电位E0
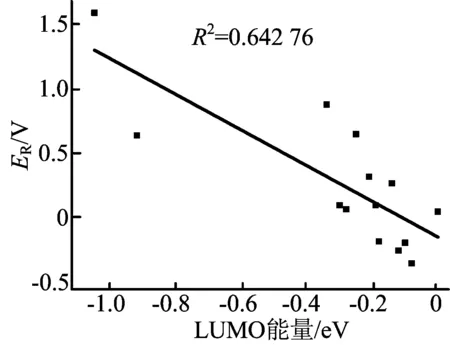
图3 LUMO能量与还原电位ER
这16个分子的HOMO能量分布在-8.95~6.32 eV之间,氧化电位在4.49~6.86 V之间,本文把HOMO能量当作自变量,氧化电位作为因变量,用Origin软件进行线性回归分析得出决定系数R2=0.943 73,回归方程斜率k=-0.99,HOMO能量与氧化电位呈显著的负相关关系。除4、12分子外的14个分子LUMO能量分布在-1.03~0.02 eV之间,还原电位在-0.35~1.59 V之间,决定系数R2=0.642 76,回归方程斜率k=-1.38,也呈显著的负相关关系。HOMO和LUMO能量与氧化还原电位线性回归分析结果符合前线轨道理论。
文献[10]通过计算32个已报道的成膜添加剂分子的锂离子结合能,发现溶剂化模型下得到的锂离子结合能均小于EC的锂离子结合能,因此认为锂离子结合能是优良成膜添加剂的重要特征。锂离子结合能(E(Li+))是中性分子(M)结合一个锂离子(Li+)后放出的吉布斯自由能。计算锂离子结合能,需要对分子和分子与Li+络合物结构分别做几何优化和频率计算,可得到两者的吉布斯自由能。
EC和苯基乙基磷酸酯(分子10)与Li+结合前后的优化结构如图4所示。图4中,箭头指向为分子的偶极矩方向,由中性分子和锂离子与该分子的锂离子络合物(Li+(M))的吉布斯自由能之差得到锂离子结合能如下:
E(Li+)=G(M)+G(Li+)-G[Li+(M)]
(7)
筛选后分子10、11、12和EC的锂离子结合能见表2所列,3个EEP同系物分子的锂离子结合能分别为0.002、0.068、0.103 eV,均低于EC的0.138 eV。
锂离子结合能的计算结果确认了苯基乙基磷酸酯(10)、苯乙烯基乙基磷酸酯(11),二苯基乙基磷酸酯(12)具有成膜添加剂的潜力。
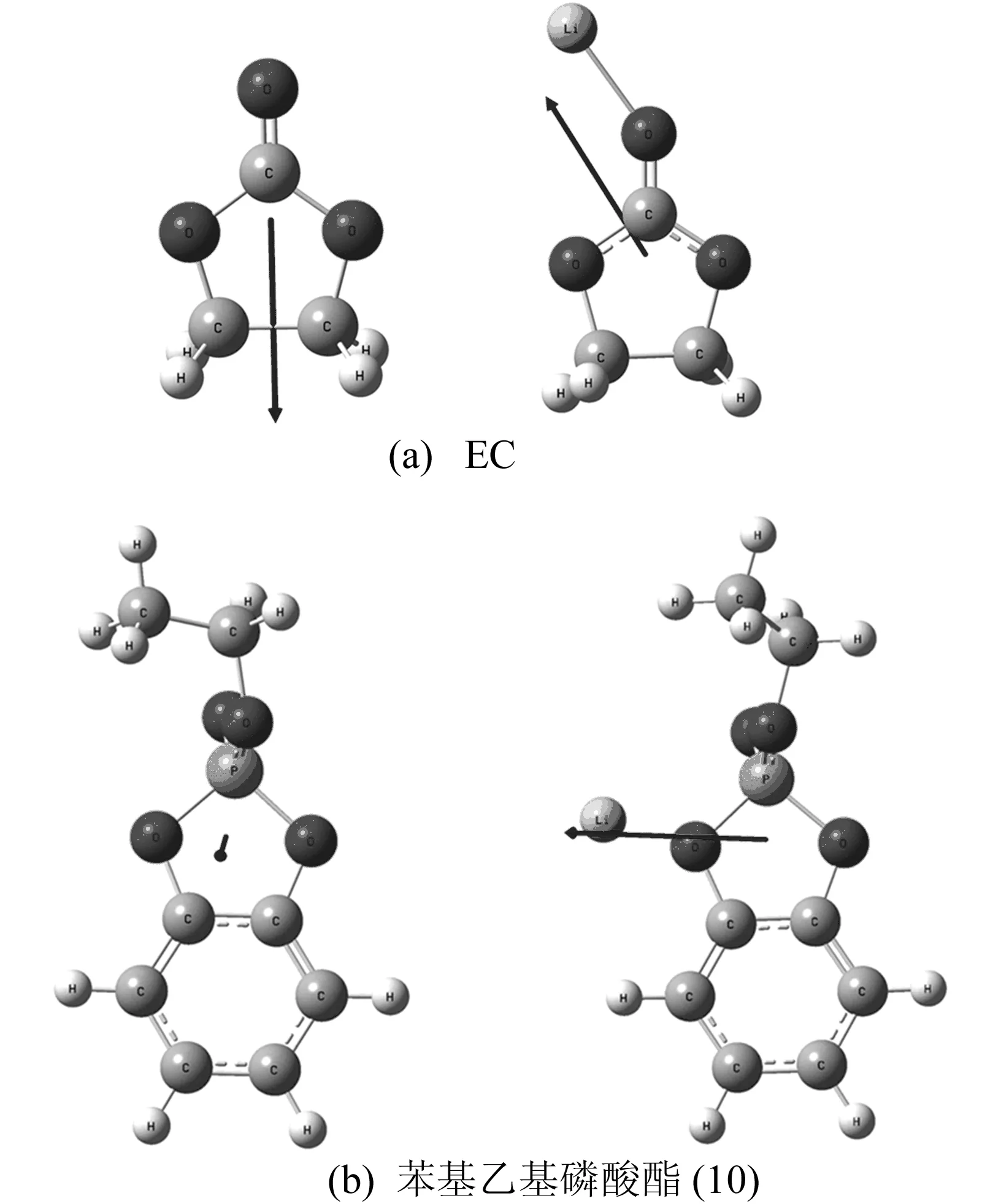
图4 分子与锂离子结合前后的优化结构

分子名称锂离子结合能/eV苯基乙基磷酸酯(10)0.002苯乙烯基乙基磷酸酯(11)0.068二苯基乙基磷酸酯(12)0.103碳酸乙烯酯(EC)0.138
4 结 论
苯基乙基磷酸酯、苯乙烯基乙基磷酸酯和二苯基乙基磷酸酯作为添加剂可能同时具有成膜和抗过充的功能。EEP同系物分子的量子化学参数有明显的官能团特征,苯环取代不仅可以提高还原电位,还可以显著降低氧化电位和化学硬度,提高分子的反应活性;氟原子取代可以提高分子的还原电位,对氧化电位和化学硬度影响并不明显。HOMO能量、LUMO能量和氧化还原电位计算结果符合前线轨道理论,但是HOMO能量与氧化电位相关度和LUMO能量与还原电位相关度的差别需要进一步明确。筛选后的分子都具有比EC更低的锂离子结合能,这进一步印证了它们具有用作锂离子电池电解液成膜添加剂的潜力。上述研究结果为环状磷酸酯作为多功能添加剂在锂离子电池电解液中的应用提供理论指导。
[1] 段飞,杜真真,杨鹏,等.锂离子电池正极材料LiMn2O4的Al2O3包覆研究[J].合肥工业大学学报(自然科学版),2013,36(3):346-351.
[2] WANG X,YASUKAWA E,KASUYA S.Nonflammable trimethyl phosphate solvent-containing electrolytes for lithium-ion batteries:I.fundamental properties[J].Journal of the Electrochemical Society,2001,148(10):A1058-A1065.
[3] OTA H,KOMINATO A,CHUN W J,et al.Effect of cyclic phosphate additive in non-flammable electrolyte[J].Journal of Power Sources,2003,119:393-398.
[4] GAO D,Xu J B,LIN M,et al.Ethylene ethyl phosphate as a multifunctional electrolyte additive for lithium-ion batteries[J].RSC Advances,2015,5(23):17566-17571.
[5] VOLLMER J M,CURTISS L A,VISSERS D R,et al.Reduction mechanisms of ethylene,propylene,and vinylethylene carbonates a quantum chemical study[J].Journal of the Electrochemical Society,2004,151(1):A178-A183.
[6] FUJIMOTO H,SATOH S.Orbital interactions and chemical hardness[J].The Journal of Physical Chemistry,1994,98(5):1436-1441.
[7] HALLS M D,TASAKI K.High-throughput quantum chemistry and virtual screening for lithium ion battery electrolyte additives[J].Journal of Power Sources,2010,195(5):1472-1478.
[8] FUKUI K,YONEZAWA T,SHINGU H.A molecular orbital theory of reactivity in aromatic hydrocarbons[J].The Journal of Chemical Physics,1952,20(4):722-725.
[9] FUKUI K,YONEZAWA T,NAGATA C,et al.Molecular orbital theory of orientation in aromatic,heteroaromatic,and other conjugated molecules[J].The Journal of Chemical Physics,1954,22(8):1433-1442.
[10] PARK M H,LEE Y S,LEE H,et al.Low Li+binding affinity:an important characteristic for additives to form solid electrolyte interphases in Li-ion batteries[J].Journal of Power Sources,2011,196(11):5109-5114.
Quantumchemicalcalculationoncyclicphosphatecompoundsasmultifunctionalelectrolyteadditives
XIANG Hongfa, LIN Min, ZHENG Hao, HE Xiaodong
(School of Materials Science and Engineering, Hefei University of Technology, Hefei 230009, China)
As multifunctional electrolyte additives for lithium-ion batteries, cyclic phosphate compounds have attracted much attention. The study of their structure-performance relationship is of importance for the composition design and application of electrolyte. In this paper, quantum chemical calculations on 15 kinds of cyclic phosphate compounds were conducted, and the frontier orbital energy, chemical hardiness and redox potentials of the cyclic phosphate compounds were obtained in order to discuss their possibility as multifunctional electrolyte additives in lithium-ion batteries. The relationship between the electrochemical stability and structures of cyclic phosphates was investigated. The results show that phenyl substitution can not only increase the reduction potential of cyclic phosphates, but also greatly reduce the oxidation potential and chemical hardiness. As a result, the reactivity of cyclic phosphates increases. Fluorine substitution can increase the reduction potential of cyclic phosphates, but has negligible effect on the oxidation potential and chemical hardiness.
lithium-ion battery; electrolyte; additive; phosphate; quantum chemistry
2016-04-14;
2016-10-19
国家自然科学基金面上资助项目(51372060);合肥工业大学2014年国家级大学生创新训练计划资助项目(201410359068)
项宏发(1981-),男,安徽无为人,博士,合肥工业大学教授,博士生导师.
10.3969/j.issn.1003-5060.2017.09.007
TM912.9
A
1003-5060(2017)09-1181-05
(责任编辑 闫杏丽)
猜你喜欢
山东冶金(2019年5期)2019-11-16
天然产物研究与开发(2018年8期)2018-09-10
中国粮油学报(2016年1期)2016-02-06
中国资源综合利用(2016年7期)2016-02-03
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01
火炸药学报(2014年5期)2014-03-20
中国粮油学报(2014年8期)2014-02-06
郑州大学学报(理学版)(2013年3期)2013-03-11
中南大学学报(自然科学版)(2012年3期)2012-07-31
农民科技培训(2009年1期)2009-02-17
