
凝胶色谱净化超高效液相色谱法测定食品中10种工业染料
2017-10-19 08:54胡海山张云伟龙洲雄
分析科学学报 2017年3期
胡海山, 鄢 兵, 张云伟, 龙洲雄
(1.江西省分析测试研究所,江西南昌 330029;2.江西省科技发展研究中心,江西南昌 330046)
工业染料具有脂溶性,经常被用在脂肪含量高的各种食品中以改善其色泽,但这些染料不但无任何营养价值,而且对人们身体健康危害极大。目前,食品中染料的传统分析方法主要有液相色谱法[1 - 3]和液相色谱-质谱联用法[4 - 7]。由于食品基质复杂,样品的前处理一般采用固相萃取[6 - 7]、分散萃取[8]、凝胶色谱[4,9 - 10]等方法进行净化。食品中工业染料检测的标准方法[11 - 12]多采用液相色谱法。这些方法大多数检测项目不多或者样品前处理过程步骤繁琐,容易造成回收率不高,不适用于多种类型食品的检测。而且随着工业染料种类的增多,食品基质越来越复杂,很多方法已无法满足检测要求,且食品中分散黄1的检测方法也未见报道。
本文以凝胶色谱(GPC)净化技术为手段,净化浓缩处理后用超高效液相色谱(UPLC)测定,建立了一个快速测定分散染料和苏丹染料等不同颜色工业染料的分析方法。方法样品处理过程简单,回收率高,检出限比普通液相色谱法低,检测更准确,适用于各种类型的食品。
1 实验部分
1.1 仪器、试剂及材料
ACQUITY UPLC H-Class型超高效液相色谱仪(美国,Waters公司),配二极管列阵检测器; Free Style型凝胶色谱净化仪(德国,LCTech公司);KQ-5200DE型超声波清洗器(昆山市超声仪器有限公司);TDL-5-A型离心机(上海安亭科学仪器厂);RE-52AA型旋转蒸发器(上海亚荣生化仪器厂);FM200型高剪切分散乳化机(德国,FLUKO公司)。
标准品:分散黄1、苏丹橙G、分散蓝3、碱性嫩黄O、分散橙1、对位红、苏丹红Ⅰ、苏丹红Ⅱ、苏丹红Ⅲ、苏丹红Ⅳ(德国Dr.Ehrenstorfer公司);甲酸(色谱纯,美国Sigma公司);乙腈、乙酸乙酯、环己烷(色谱纯,美国Fisher公司);超纯水由Milli-Q Direct8 超纯水机(默克密理博)制备。
样品:市场购买。
1.2 实验方法
1.2.1标准溶液配制标准储备溶液:准确称取碱性嫩黄O、分散黄1、苏丹橙G、分散蓝3、对位红、分散橙1、苏丹红Ⅰ、苏丹红Ⅱ、苏丹红Ⅲ、苏丹红Ⅳ各约5 mg(精确到0.01 mg),分别置于25 mL容量瓶中,用乙腈超声溶解并定容至刻度,于4 ℃冰箱中保存。混合标准溶液:准确吸取各标准储备液1 mL于25 mL容量瓶中,用乙腈定容,于4 ℃冰箱中保存,备用。混合标准系列溶液:准确吸取10种染料混合标准溶液0.02、0.05、0.1、0.2、0.3、0.5 mL,用乙腈定容至1 mL,分别配制成浓度约为0.16、0.4、0.8、1.6、2.4、4.0 μg/mL的混合标准系列进行线性测定,临用时现配。
1.2.2凝胶色谱分离净化条件色谱柱:Bio-Beads S-X3 净化柱(500×25 mm),流动相:环己烷-乙酸乙酯(1∶1,V/V);流速:5 mL /min;进样量:5 mL;流出液收集时间:16~32 min。
1.2.3超高效液相色谱测定条件色谱柱:Waters ACQUITY UPLC®BEH C18柱(100×2.1 mm,1.7 μm);流动相A相为乙腈,B相为0.1%甲酸;梯度洗脱程序为:0~4.0 min,35~60%A;4.0~5.0 min,60~80%A;5.0~9.0 min,80%A;9.0~9.1 min,80~100%A;9.1~15.0 min,100%A;流速0.2 mL/min;柱温30 ℃;进样体积:3 μL;检测波长:254 nm。
1.2.4样品处理食用油样品:称取约2 g(准确至0.0001 g)样品于小烧杯中,加入适量的环己烷-乙酸乙酯(1∶1,V/V)溶解,难溶解的样品可加温溶解,最后定容至10 mL。过滤膜后按照1.2.2项下凝胶色谱条件净化,收集的流出液经旋转蒸发至近干,再用氮气吹干,最后用乙腈定容至1 mL,微膜过滤后上机测定。卤肉、油炸食品、火锅底料等样品:称取约5 g(准确至0.0001 g)样品于离心管中,加10 mL环己烷-乙酸乙酯(1∶1,V/V)后将其匀浆分散,超声震荡15 min后,4 000 r/min离心10 min,收集上清液,再往离心管中加入5~10 mL环己烷-乙酸乙酯(1∶1,V/V),超声震荡15 min后,4 000 r/min离心10 min,合并上清液,提取液用氮吹仪氮气吹干至10 mL以下,最后用环己烷-乙酸乙酯(1∶1,V/V)定容至10 mL。过滤膜后按照1.2.2项下凝胶色谱条件净化后,收集的流出液经旋转蒸发至近干,再用氮气吹干,最后用乙腈定容至1 mL,微膜过滤后上机测定。
2 结果与讨论
2.1 样品提取溶剂的确定
染料化合物均为脂溶性,根据相似相溶原理,试验了乙酸乙酯、甲醇、乙腈、环己烷几种常见的有机溶剂,对实际样品提取效果进行了比较。结果发现甲醇和乙腈与乙酸乙酯和环己烷对脂肪含量较高的样品提取效果存在一定差别,甲醇和乙腈提取的回收率偏低。考虑到对样品中脂肪的溶解性和凝胶色谱净化过程使用的流动相是乙酸乙酯-环己烷(1∶1,V/V),为了避免溶剂替换过程中目标物的损失和不同极性提取液对凝胶色谱柱溶胀度的影响,本实验采用乙酸乙酯-环己烷(1∶1,V/V)作为提取液。
2.2 超高效液相色谱测定条件的确定
实验选择10种具有不同颜色的染料作为检测组分,但各组分的最灵敏检测波长差别较大,因此选择了210~600 nm全波长检测。结果显示,在254 nm下均有吸收且灵敏度较好,10种染料除了在紫外波长下有吸收外,在可见光波长区也均有特征吸收,碱性嫩黄O、分散黄1、苏丹橙G在370 nm左右,分散蓝3在594 nm,对位红、分散橙1、苏丹红Ⅰ、苏丹红Ⅱ在480 nm左右,苏丹红Ⅲ、苏丹红Ⅳ在510 nm左右,有的组分在可见光区特征吸收比254 nm的灵敏度高达2倍。在实际样品检测时,卤肉和火锅底料在254 nm下存在杂质吸收峰干扰,很多干扰物质在可见光波长区域几乎没有吸收,背景干扰小,基线稳定。为了判断杂质干扰和获得更高的灵敏度,可选择210~600 nm全波长检测。
工业染料是疏水性物质,乙腈洗脱强度大,而且对工业染料的溶解性好,因此本实验采用乙腈作为流动相的有机相。实验分别采用乙腈-水、乙腈-0.02 mol/L 乙酸铵、乙腈-0.1%甲酸三种不同流动相进行对比,结果发现乙腈-0.1%甲酸作为流动相,不但峰形好,而且分离度最佳,见图1。
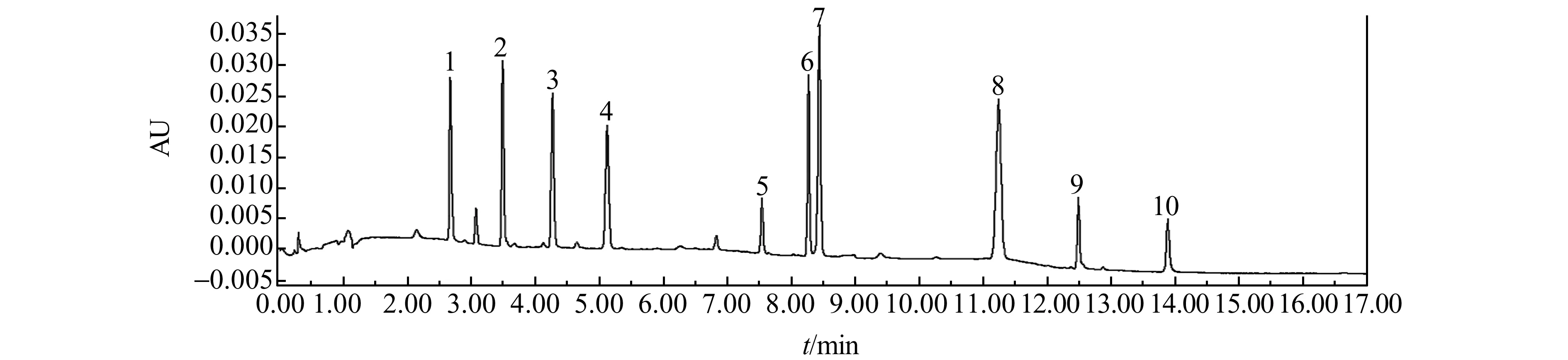
图1 10种染料混合标准的色谱图(乙腈-0.1%甲酸)Fig.1 Chromatograms of mixed standards of 10 dyes (acetonitrile -0.1% formic acid)1.Auramine O;2.Disperse yellow 1;3.Sudan orange G;4.Disperse blue 3 5.Para red;6.Disperse orange 1;7.Sudan red Ⅰ;8.Sudan redⅡ;9.Sudan red Ⅲ;10.Sudan red Ⅳ.
2.3 凝胶色谱分离净化条件的确定
通常染料检测的样品基质较为复杂,一般含有较多的脂肪、蛋白质等大分子杂质,而凝胶色谱法可以根据分子量大小来有效分离杂质.本实验采用凝胶色谱为净化手段,充分考虑对样品中被测组分的溶解性来进行净化处理。实验采用10种染料的混合标准溶液进样,分别在进样后5、10、15、20、25、30 min开始收集流出液,收集至35 min,浓缩后用超高效液相色谱法测定,发现只有15、20和25 min开始收集的流出液含有染料组分,为了确保充分收集被测组分,因此本实验确定收集流出液时间为16~32 min。
实验比较了样品不经过凝胶色谱和经过凝胶色谱的净化效果,分别见图2、图3。从图中可以看出,图2中几个大的杂质峰在图3中都未出现,表明凝胶色谱净化过程能有效地去除样品中部分杂质。样品经过凝胶色谱净化,浓缩吹干后,也明显看到旋转蒸发玻璃瓶壁上没有附着脂肪,样品中的脂肪都被去除。结果显示,凝胶色谱净化过程能有效分离部分杂质和脂肪。
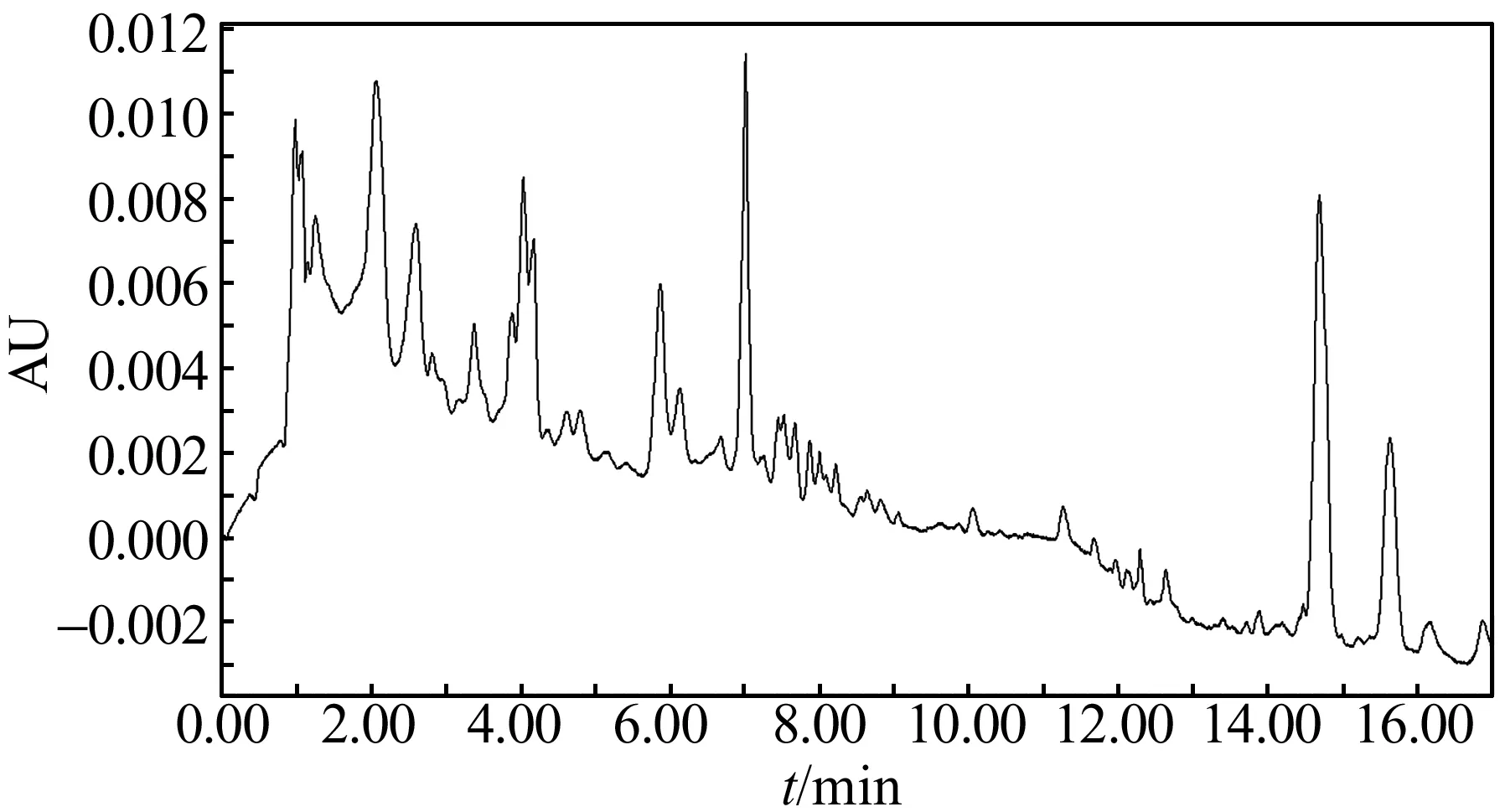
图2 未经过凝胶色谱净化处理酱卤肉样品的色谱图(λ=254 nm)Fig.2 Chromatograms of sauced meat sample without purification by gel permeation chromatography(λ=254 nm)

图3 经过凝胶色谱净化处理的酱卤肉样品的色谱图(λ=254 nm)Fig.3 Chromatograms of sauced meat sample purified by gel permeation chromatography(λ=254 nm)
2.4 方法学考察
2.4.1方法的标准曲线及检出限取1.2.1中混合标准系列溶液上机测定,以待测化合物色谱峰面积为纵坐标(Y),相应目标物浓度为横坐标(X)进行线性回归分析,得到线性回归方程及相关系数。以信噪比(S/N>3)计算,得出10种染料的检出限。结果见表1。

表1 10种染料的线性方程、相关系数和检出限
2.4.2方法的回收率和精密度取不含染料的酱卤肉作为空白样品,分别加入高、中、低三个浓度的染料混合标准溶液,按1.2.4项下的样品前处理方法处理样品后上机测定,每个加标水平平行测定5次。由表2可知,10种染料的平均回收率为65.0%~107.2%,相对标准偏差(RSD)为2.60%~6.66%。本方法具有良好的精密度,10个组分中只有碱性嫩黄O和分散蓝3回收率偏低。
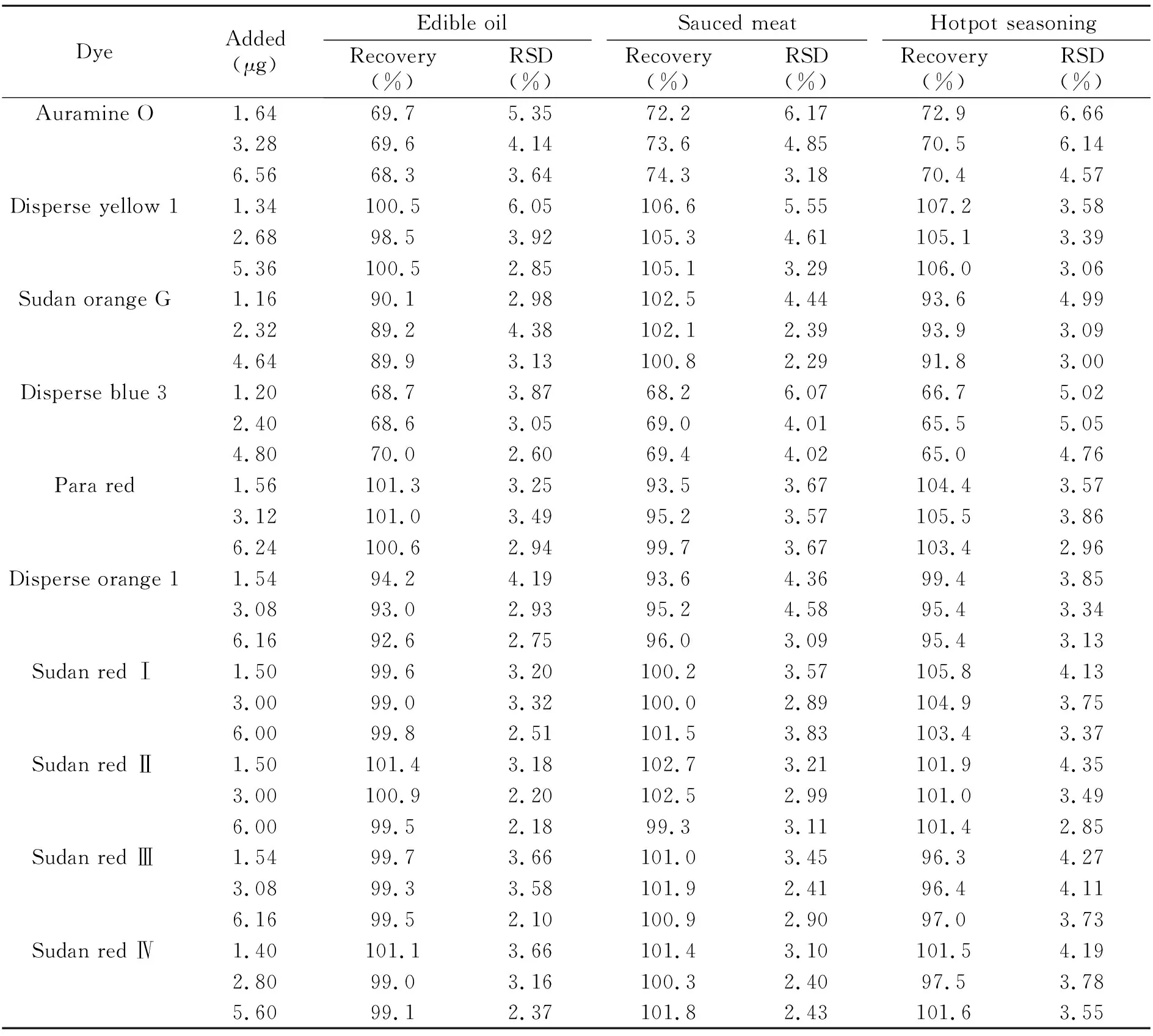
表2 10种染料的加标回收率和相对标准偏差(n=5)
2.4.3稳定性试验为了考察10种染料在实际样品处理后的稳定性,取2.4.2项下的高、中、低三个空白样品加标处理后的样品溶液,按1.2.3项实验条件,每隔2 h、4 h、8 h、1 d和1 w进行实验,观察10种染料的峰面积变化情况。结果发现,处理后放在冰箱内4 ℃冷藏的样品溶液中10个组分基本很稳定,一个星期内未见明显变化,各染料组分峰面积变化RSD≤2.58%,稳定性较好。
2.4.4样品测定结果按实验方法对市场销售的5个食用油、5个酱卤菜、5个火锅底料、5个油炸食品进行了测定,样品中均未检出染料成分。
3 结论
利用凝胶色谱法净化,建立了食品中10种工业染料的超高效液相色谱测定法,并确定了提取净化条件和超高效液相色谱测定条件。方法不但样品处理过程简单,而且有较好的灵敏度、回收率和精密度,可满足食品中10种染料的检测要求。方法处理快速高效,苏丹红的检测时间比国家标准缩短了25 min,在多组分快速检测方面有明显优势。
猜你喜欢
昆明医科大学学报(2021年8期)2021-08-13
化工管理(2021年7期)2021-05-13
食品安全导刊(2020年14期)2020-12-04
环境保护与循环经济(2020年4期)2020-06-08
中国特种设备安全(2019年1期)2019-03-13
现代食品(2018年16期)2018-11-02
中成药(2018年6期)2018-07-11
健康博览(2017年12期)2018-02-06
中成药(2017年8期)2017-11-22
食品工业科技(2016年17期)2016-10-31
