
不对称双席夫碱Ni配合物催化环己烯环氧化
2017-10-13 01:28朱路群胡春梅吕兴强
化工技术与开发 2017年9期
王 静,卢 荣,朱路群,胡春梅,吕兴强
(1.西北大学化工学院,陕西 西安 710069;2.西安雅芝生物科技有限公司,陕西 西安 710000)
不对称双席夫碱Ni配合物催化环己烯环氧化
王 静1,2,卢 荣1,朱路群1,胡春梅1,2,吕兴强1
(1.西北大学化工学院,陕西 西安 710069;2.西安雅芝生物科技有限公司,陕西 西安 710000)
本文以3,5-二叔丁基水杨醛与邻苯二胺合成席夫碱配体(L2),用该配体与Ni2+配位合成中间体(L2-Ni),再与不同的芳香醛反应制得一系列不对称Salen-Ni(Ⅱ)配合物。利用傅里叶变换红外光谱和元素分析对配体和配合物进行了表征。然后以30%的过氧化氢为氧源,以一系列不对称Salen-Ni(Ⅱ)配合物为催化剂,探究了环己烯环氧化反应的催化性能和配体上不同推拉电子基团对催化反应的影响。通过优化反应条件,环己烯的转化率和选择性最高可达到67.4%、43.5%。
不对称Salen-Ni(Ⅱ);环己烯;环氧化反应;过氧化氢
环氧环己烷是一种重要的有机反应中间体,其分子结构中存在活泼的环氧基团,可以发生水解、醇解、胺解得到1,2-环己二醇、邻烷氧基环己醇、邻氨基环己醇,也可以和CO2发生开环聚合生成聚碳酸酯[1]等,所以环氧环己烷的制备是科学领域研究的重点之一。
环氧环己烷主要以环己烯为原料催化环氧化而得。在催化氧化环己烯的过程中,使用的氧源种类较多,有过氧酸[2]、TBHP[3]、亚碘酰苯[4]、次氯酸钠[5]、过氧化氢等。以过氧化氢为氧源时,其产物是水,对环境没有污染,是一种环境友好型氧化剂。催化氧化环己烯制备环氧环己烷的热点仍是催化剂的研究与利用,目前已经有多种催化环氧化环己烯的催化剂。席夫碱过渡金属配合物因具有良好的催化性能而被广泛应用于环己烯环氧化中。过渡金属配合物的中心离子主要有 Ru、Mn、Cu、Ni、Fe、Co、Cr等,此类催化剂配体主要以对称的席夫碱为主,而以非对称席夫碱作为配体的报道较少[1]。
针对以上问题,本文设计一系列不对称Salen-Ni(Ⅱ)配合物,通过改变配体上的推拉电子基团来调节中心离子的电子效应,从而实现对催化剂的催化性能调控。配体的电子给予或接受能力会影响中心离子的电子效应,使得配合物的电子云密度发生改变,导致配合物的化学性质发生变化。本文以环己烯为原料,过氧化氢为氧化剂,考察了该系列配合物作为催化剂,催化氧化环己烯的催化性能,并且探究了非对称席夫碱配体上的推拉电子基团对中心离子的催化性能的影响。
1 实验部分
1.1 主要试剂和仪器
环己烯(GR),30% 过氧化氢(AR),邻苯二胺(AR),3,5-二叔丁基水杨醛(AR),5-溴水杨醛(AR),邻香草醛(AR),水杨醛(AR),3,5-二溴水杨醛(AR),5-溴邻香草醛(AR),乙腈、乙醇、三氯甲烷、乙酸乙酯、甲醇等(均为分析纯)。
FT-IR光谱分析仪,VarioEL Ⅲ型元素分析仪,GC2060气相色谱仪,氢火焰离子化检测器(载气为N2,色谱柱为SE-30,检测反应时,柱温100℃,检测器和汽化室分别为220℃和230℃)。
1.2 催化剂的合成
1.2.1 配体(L2)的合成
根据文献[6],在50mL圆底烧瓶中加入2.1mmol邻苯二胺、20mL乙醇,然后再加入3,5-二叔丁基水杨醛1mmol,再加入10mL乙醇,加热回流4h。反应结束后,产物在甲醇中重结晶,得到橘黄色配体L2,反应式如Scheme 1所示。Anal.calcd for C21H28N2O:C 77.74,H 8.70,N 8.63;found:C 77.64,H 8.41,N 8.75。

Scheme 1 配体L2的合成路线Scheme 1 Synthetic route for ligand L2
1.2.2 不对称Salen-Ni(Ⅱ)配合物的合成
在50mL烧瓶中,取0.025mmol的L2配体溶于3mL乙醇,然后加入0.025 mmol醋酸镍加热回流0.5h,冷却至室温。再加入0.01mmol芳香醛(a:邻香草醛,b:5-溴邻香草醛,c:3,5-二溴水杨醛,d:5-溴水杨醛,e:水杨醛),分别加入1mL乙醇、1mL二氯甲烷,常温搅拌1h。过滤,收集滤液,等待溶剂挥发,收集产物。反应式如Scheme 2所示。Anal.calcd for C29H30Cl2N2NiO3:(a) C 59.63,H 5.18,N 4.80;found C 59.23,H 5.01,N 4.92。Anal.calcd for C29H29BrCl2N2NiO3:(b) C 52.53,H 4.41,N 4.22;found C 52.23,H 4.35,N 4.51。Anal.calcd for C28H26Br2Cl2N2NiO2:(c) C 47.24,H 3.68,N 3.93;found C 47.01,H 3.81,N 3.79。Anal.calcd for C28H27BrCl2N2NiO2:(d) C 53.13,H 4.30,N 4.43;found C 52.98,H 4.41,N 4.31。Anal.calcd for C28H28Cl2N2NiO2:(e) C 60.69,H 5.09,N 5.06;found C 60.51,H 5.01,N 5.18。
1.3 催化环己烯环氧化
在50mL圆底烧瓶中加入0.01mmol不对称Salen-Ni催化剂、2mL 乙腈、1.5mol·L-1的碳酸氢钠溶液2mL和30%的过氧化氢溶液0.06mmol,混合均匀后,再加入2mmol反应底物,常温搅拌2h。反应结束后,用三氯甲烷萃取,静置分层,分出下层有机相。用无水硫酸钠进行干燥,然后定容至10mL。
用气相色谱对产物进行分析,通过外标法进行计算,分析结果为:环己烯的保留时间1.84min,环氧环己烷2.723min,乙腈1.398min,三氯甲烷1.548min。反应式如Scheme 3所示。

Scheme 2 Salen-Ni(Ⅱ)配合物的合成路线Scheme 2 Synthetic route for Salen-Ni(Ⅱ)complexes

Scheme 3 环己烯氧化反应示意图Scheme 3 Possible process for the oxidation of cyclohexene
2 结果与讨论
2.1 催化剂的合成原理
合成的配体(L2)在有机相中与Ni盐的醇溶液进行反应,Ni离子很容易与配体L2进行单边配位,形成中间体L2-Ni。随后再加入芳香醛类,使得Ni达到配位平衡,制备出不对称双席夫碱Ni催化剂。由于芳香醛上的推拉电子基团不同,可实现对中心离子Ni上电子云的密度调节,进而调节催化剂的催化活性。催化剂的结构如Scheme 4所示。

Scheme 4 催化剂的结构Scheme 4 The Structure of the catalyst
2.2 催化剂的表征
图1~图3所示为配体(L2)的FT-IR图谱。在3487cm-1处出现了NH2的伸缩振动吸收带;配体在2866cm-1处出现宽带弱吸收峰,是酚羟基(O-H)的伸缩振动。由于OH…N形成分子内氢键,所以向低频方向移动。1268cm-1处中等强度吸收峰是O-H的变性振动。1615cm-1处出现强吸收峰,是配体(L2)特征基团-C=N-键的伸缩振动吸收峰[7]。
催化剂a:金属Ni与配体和邻香草醛配位后,O-H在1268cm-1和2866 cm-1处出现的吸收峰都消失了,说明O配位后脱去一个质子。配合物和配体(L2)相比,C=N键的伸缩振动峰发生了红移,说明N与Ni发生了配位。并且在3500 cm-1~3250cm-1处的NH2的中等强度吸收峰消失,在1248cm-1和1113cm-1处出现了苯环上OCH3的伸缩振动吸收峰。
催化剂b:在1240cm-1和1182cm-1处的中强吸收峰是苯环上OCH3的伸缩振动峰,993cm-1处的中强吸收峰是C-Br的伸缩振动带。
催化剂c:除了以上特征,在1131cm-1和532cm-1处出现了C-Br的伸缩振动带,说明在分子式中存在两个溴原子。
催化剂d:与催化剂c相比,只在1131cm-1处出现了C-Br伸缩,说明只有一个溴原子。
催化剂e:金属Ni与配体和水杨醛配位后,O-H在1268cm-1和2866cm-1处出现的吸收峰消失,说明O配位后脱去一个质子。配合物和配体(L2)相比,C=N键的伸缩振动峰发生了红移,说明N与Ni发生了配位;并且在3500cm-1~3250cm-1处的NH2的中等强度吸收峰消失。

图1 红外光谱图Fig.1 FT-IR spectra of (1) ligand L2, (2) catalyst e
2.3 催化反应结果
用制备的不对称Salen-Ni(Ⅱ)为催化剂催化环己烯环氧化反应,产物均用GC 2060气相色谱仪定量分析,氢火焰离子化检测器,SE-30型毛细管色谱柱,N2为载气,柱温、气化室和检测器的温度分别是 130℃、240℃、240℃。采用外标法进行计算,转化率和选择性的计算方法如式(1)和式(2)[8]:
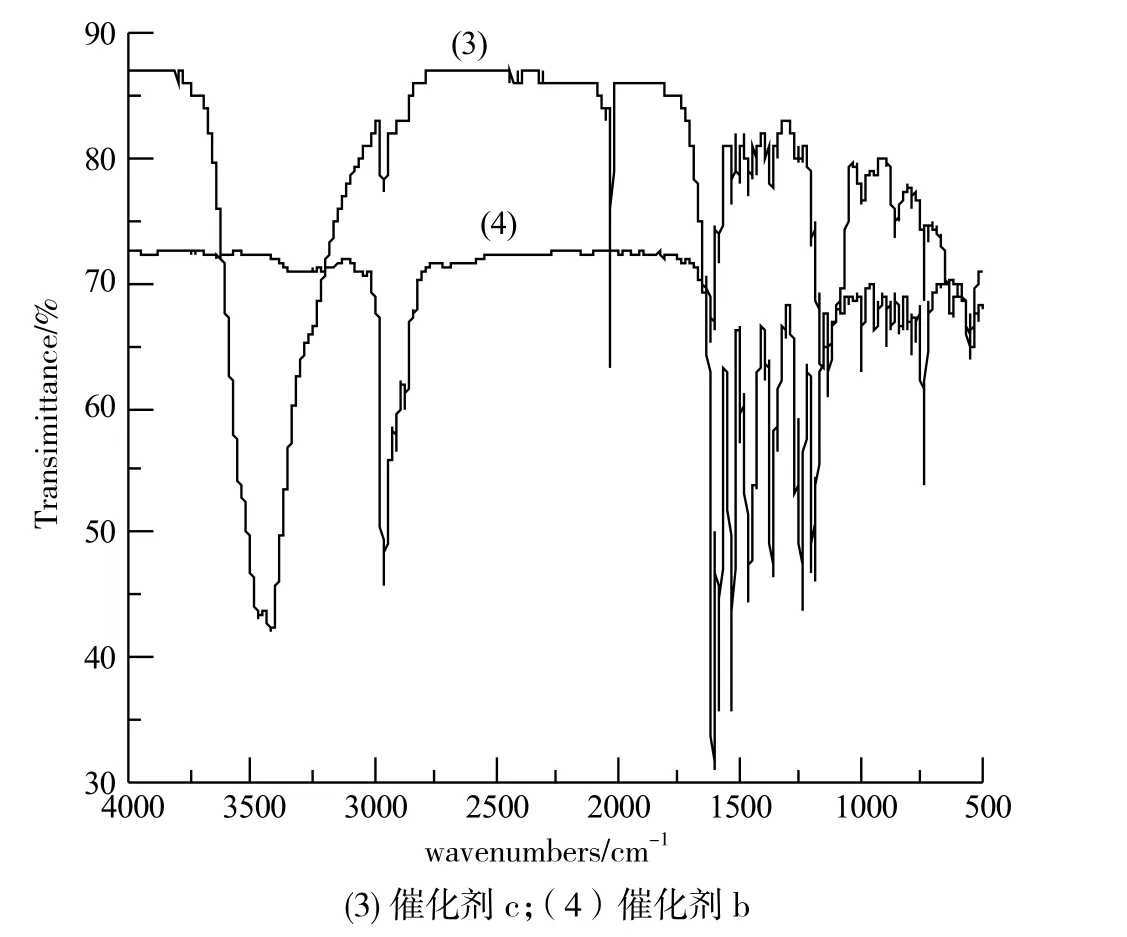
图2 红外光谱图Fig.2 FT-IR spectra of (3) catalyst c (4) catalyst b

图3 红外光谱图Fig.3 FT-IR spectra of (5) catalyst d (6) catalyst a

由于5种催化剂的结构不同,其催化效果也应该有一定的差异,因为配体上的推拉电子基团和空间结构不同,配合物中心离子的路易斯酸性不同。因此,我们先以配合物d(L2-Ni-5-溴水杨醛)为催化剂,进行一系列单因素优化。然后再以最优条件对其他催化剂进行拓展,进一步比较配体上的推拉电子基团对催化剂的催化活性影响。
2.3.1 催化剂用量对反应影响
催化剂用量对反应的影响见图4。催化剂用量增加到0.01mmol的过程中,反应速率和选择性增快。然而,继续增加催化剂的用量,环己烯的转化率和环氧环己烷的选择性反而下降。这可能与典型的金属卟啉催化反应的特点相似,即催化活性中间体的量增加引起的屏蔽效应,使底物与催化剂中间体接触的机率减少[9],从而环氧环己烷的转化率和选择性降低。

图4 催化剂用量对反应的影响Fig.4 The effect of catalyst amount on reaction
2.3.2 温度对反应的影响
图5为温度对环己烯环氧化反应的影响。由图5可知,温度对反应有很大的影响,在低温条件下,环己烯环氧化反应很难进行,环己烯转化率和环氧环己烷的选择性只有19.1%和9.2%。随着温度的升高,环己烯的转化率和环氧环己烷的选择性迅速增加。这是因为温度是影响速率常数(k)的主要因素,反应速率会随着温度的升高而增加。当温度达到35℃以上,环己烯的转化率基本保持不变,而环氧环己烷的选择性呈现降低的趋势。根据反应机理推测,在较高温度下,生成的环氧环己烷发生开环氧化反应,导致环氧环己烷的选择性降低[10]。

图5 温度对反应的影响Fig.5 The effect of temperature on reaction
2.3.3 时间对反应的影响
图6为时间对环己烯环氧化反应的影响。由实验结果可知,随着反应时间的延长,环己烯的转化率逐渐增高。而选择性的增加幅度相对较大,6h左右选择性增加至43.5%。主要原因是在反应前几个小时,催化剂跟氧源生成氧化中间产物的量较多,活性中间体与环己烯碰撞机率较大,因此环氧环己烷在前几小时的选择性较高[11]。但是,反应时间过长,环氧环己烷发生开环分解,生成相应的副产物而使得环氧环己烷的选择性降低,因此,时间在6h左右反应效果比较好。

图6 时间对反应的影响Fig.6 The effect of time on reaction
2.3.4 过氧化氢/环己烯的比例对反应的影响
图7为环己烯/过氧化氢的比例对环氧化反应的影响。随着氧源比例的逐渐增加,环己烯的转化率从65%增加到86%。而环氧环己烷的选择性先增高而后大幅度降低,可能原因是在氧源从1∶1增加到1∶2的过程中,氧源/催化剂的比例较合适,活性中间体浓度较高,环氧环己烷产率不断增加。但是,氧源比例增加,催化剂传递“氧”的活性达到最大值,氧源相对过量,氧源直接作用于底物,副反应加剧[12],选择性出现下降的趋势。因此,氧源比例选择为1∶2较合适。

图7 氧源比例对反应的影响Fig.7 The effect of oxidation ratio on reaction
2.3.5 溶剂对反应的影响
溶剂的选择在反应中起到重要的作用[13],本文研究了乙腈、三氯甲烷、甲醇、乙酸乙酯这4种不同的溶剂对环氧化反应的影响,反应结果如表1所示。在极性非质子性溶剂乙腈中,环己烯的转化率和选择性较高,选择性和转化率达到65.7%和29.2%。可能是乙腈作溶剂时反应体系为均相体系,氧源很容易和底物接触,可以提供较好的反应环境。在溶剂乙醇和乙酸乙酯中,环氧化产物的选择性几乎为零,是由于反应过程中生成的环氧化产物很容易跟其发生开环反应[14]。在三氯甲烷中,反应体系为非均相,转化率很低。因此,乙腈为反应的最佳溶剂[15]。

表1 溶剂单因素Table 1 The single-factors of solvent
2.4 不同催化剂的催化效果
通过上述优化,在最优条件下用一系列不对称Salen-Ni催化剂进行催化环己烯环氧化反应,反应结果见表2。
从表2分析可知,配体上的推拉电子基团对转化率有较强的影响,拉电子能力越强,转化率越高。如L2-Ni-3,5-二溴水杨醛上有2个拉电子基团,使得环己烯的转化率高达81.9%。环氧环己烷的选择性跟中心离子的路易斯酸性也有一定的联系,酸性过强或过弱,都会导致环氧环己烷的选择性降低。例如3,5-二溴水杨醛的选择性较5-溴水杨醛低一些, L2-Ni-邻香草醛的选择性仅9.1%,原因可能是中心离子酸性过强,生成的活性中间体容易吸附环氧环己烷,发生深度氧化。中心离子的酸性过弱,活性中间体形成比较困难,对环氧化产物的选择性较差[16]。从实验结果得知,催化剂L2-Ni-5-溴水杨醛中心离子的酸性适中,因此具有良好的催化性能。

表2 不同催化剂反应结果Table 2 The influence of different catalysts
3 结论
不对称Salen-Ni(Ⅱ)配合物对环己烯环氧化反应有较高的催化活性和选择性,并且反应条件温和。通过优化得到的最优条件是:催化剂用量0.01mmol,CH3CN 2mL,时间 6h,氧源比例 1∶2,环己烯的转化率和选择性分别达到67.4%、43.5%。由实验结果发现,配体上的推拉电子基团对催化环氧化反应有较大的影响,L2-Ni-5-溴水杨醛的催化活性最高。由于5-溴水杨醛对路易斯酸中心离子的酸性调节适中,其催化效果较好。
[1] KOJI N, MITSURU N, KYOKO N. Alternating copolymerization of cyclohexene oxide with carbon dioxide catalyzed by (salalen)CrCl complexes [J]. Macromolecules, 2009, 42 (18): 6972-6980.
[2] 许胜,陈英,胡国军.丁二烯副产物4-乙烯基环己烯的 1,2-环氧化反应 [J].石油化工,2007,36(2):169-172.
[3] MASOUD S N, SEYED N M. Synthesis, characterization and catalytic oxyfunctionalization of cyclohexene with tertbutylhydroperoxide and hydrogen peroxide in the presence of alumina-supported Mn(Ⅱ), Co(Ⅱ), Ni(Ⅱ) and Cu(Ⅱ)bis(2-hydroxyanil)benzil complexes [J].Journal of Molecular Catalysis A: Chemical, 2007, 268(1/2): 50-58.
[4] AGARWAL D D, RAJEEV J , ABHIJIT C, et al. Oxidation of alkenes using the RuCl3/PhIO system [J]. Polyhedron, 1992,11(4): 463-467.
[5] DEBABRATA C, SANGHAMITRA M, ANANNYA M.Epoxidation of olefins with sodium hypochloride catalysed by new Nickel(Ⅱ)-Schiff base complexes [J]. Journal of Molecular Catalysis A: Chemical, 2000, 154(1): 5-8.
[6] MUNOZ-HERNANDEZ M. A, KEIZER T S, PARKIN S, et al. Group 13 Cation formation with a potentially tridentate ligand [J]. Organometallics, 2000, 19(21): 4416-4421.
[7] 张萍,杨梅,吕效平.Salen Co(Ⅱ)配合物催化苯乙烯的环氧化研究[J].分子催化,2007,21(1):48-53.
[8] CUI H, ZHANG Y, QIU Z G. Synthesis and characterization of cobalt-substituted SBA-15 and its high activity in epoxidation of styrene with molecular oxygen [J]. Applied Catalysis B: Environmental, 2010, 101(1/2): 45-53.
[9] GUO, C C, LIU Q, WANG X T, et al. Selective liquid phase oxidation of toluene with air [J]. Applied Catalysis A:General, 2005, 282(1/2): 55-59.
[10] GURU B B V, SWAGATIKA S, KULAMANI P. La Complex@Fe-PILM offering resilient option for efficient and green processing toward epoxidation of cyclohexene[J].Industrial And Engineering Chemistry Research, 2011,50(15): 8973-8982.
[11] JIANG J, MA K, ZHENG Y F, et al. Cobalt salophen complex immobilized into montmorillonite as catalyst for the epoxidation of cyclohexene by air [J].Applied Clay Science, 2009, 45(3): 117-122.
[12] ZHOU X T, JI H B. Biomimetic kinetics and mechanism of cyclohexene epoxidation catalyzed by metalloporphyrins [J].Chemical Engineering Journal, 2010, 156(2): 411-417.
[13] CLERICI M G, BELLUSSI G, ROMANO U. Synthesis of propylene oxide from propylene and hydrogen peroxide catalyzed by titanium silicalite [J]. Journal of Catalysis,1991, 129(1): 159-167.
[14] FAN W B,WU P,TATSUMI T. Unique solvent effect of microporous crystalline titanosilicates in the oxidation of 1-hexene and cyclohexene [J]. Journal of Catalysis, 2008,256(1): 62-73.
[15] DING Y , GAO Q , Li G X , et al. Selective epoxidation of cyclohexene to cyclohexene oxide catalyzed by Keggintype heteropoly compounds using anhydrous ureahydrogen peroxide as oxidizing reagent and acetonitrile as the solvent [J]. Journal of Molecular Catalysis A:Chemical,2004, 218(2): 161-170.
[16] 孙伟,夏春谷.手性金属Salen配合物在不催称催化中的应用[J].化工进展,2002,14(1): 9-16.
Abstract:Ligand L2 was synthetized with 3,5-Di-tert-butyl-2-hydroxybenzaldehyde and o-phenylenediamin, which was coordinated with Ni2+. A series of asymmetric Salen-Ni (Ⅱ) complexes were synthetized by different aromatic aldehyde and L2-Ni.The complexes and ligand L2 were characterised by FT-IR, elemental analysis. The in fl uence of the catalytic properties and different electron-releasing groups or electron-withdrawing groups on ligand of the epoxidation of cyclohexene were explored with 30% H2O2as oxidation and a series of asymmetric Salen-Ni(Ⅱ) complexes as catalysts. The result showed the conversion and selectivity of cyclohexene attained to 67.4% and 43.5% under the optimum reactive conditions.
Key words:asymmetric salen-Ni(Ⅱ); cyclohexene; epoxidation reaction; hydrogen peroxide
Cyclohexene of Styrene by Novel Asymmetric Schiff base Copper Complex
WANG Jing1,2, LU Rong1, ZHU Luqun1, HU Chunmei1,2, LYU Xingqiang1
(1. School of Chemical Engineering, Northwest University, Xi’an 710069, China; 2.Xi’an Yazhi Biological Technology Co. Ltd.,Xi’an 710000, China)
O 643
A
1671-9905(2017)09-0005-06
陕西省教育厅专项科研计划项目(12JK0577)
王静(1983-),女,陕西西安人,工程师,主要从事化学工程与工艺等研究。E-mail: chemwj@163.com
卢荣(1960-),女,工程师,教授,E-mail: lu78441@aliyun.com
2017-06-05
猜你喜欢
大电机技术(2022年5期)2022-11-17
科学导报(2022年28期)2022-05-24
化工管理(2021年7期)2021-05-13
化工管理(2021年3期)2021-01-29
化工管理(2020年26期)2020-10-09
环境保护与循环经济(2020年4期)2020-06-08
世界农药(2019年4期)2019-12-30
中国特种设备安全(2019年1期)2019-03-13
石油化工技术与经济(2019年3期)2019-02-13
筑路机械与施工机械化(2017年6期)2017-07-10
