
高pH环境对胶体在饱和多孔介质中迁移的影响
2017-09-25 07:05:44袁瑞强王仕琴山西大学环境与资源学院山西太原00006山西大学资源与环境工程研究所山西太原00006江西师范大学鄱阳湖湿地与流域研究教育部重点实验室江西南昌0022中国科学院遗传所农业资源研究中心中国科学院农业资源重点实验室河北省节水农业重点实验室河北石家庄050021
中国环境科学 2017年9期
袁瑞强,郭 威,王 鹏,王仕琴(1.山西大学环境与资源学院,山西 太原 00006;2.山西大学资源与环境工程研究所,山西 太原 00006;.江西师范大学鄱阳湖湿地与流域研究教育部重点实验室,江西 南昌 0022;.中国科学院遗传所农业资源研究中心,中国科学院农业资源重点实验室,河北省节水农业重点实验室,河北 石家庄050021)
高pH环境对胶体在饱和多孔介质中迁移的影响
袁瑞强1*,郭 威1,2,王 鹏3,王仕琴4(1.山西大学环境与资源学院,山西 太原 030006;2.山西大学资源与环境工程研究所,山西 太原 030006;3.江西师范大学鄱阳湖湿地与流域研究教育部重点实验室,江西 南昌 330022;4.中国科学院遗传所农业资源研究中心,中国科学院农业资源重点实验室,河北省节水农业重点实验室,河北 石家庄050021)
以超纯水漂洗和经酸处理过的玻璃微珠作为对照,通过比较在不同pH环境下胶体在两种多孔介质中的吸附解吸行为来探讨高pH环境对胶体迁移的影响. 结果表明,不利条件下通过提高环境的pH可以使吸附在初级势阱中的胶体解吸下来,从而降低胶体的吸附能力. 不利条件下化学杂质有利于胶体在初级势阱中的吸附. 然而随着环境pH的增加,这些因化学杂质而发生吸附的胶体会有部分被释放出来. 当溶液的pH增加到 10或更高后,化学杂质对胶体吸附过程的影响可以忽略. 胶体在多孔介质表面的吸附数量与胶体的吸附解吸过程和溶液的环境有关,尤其是高pH值环境. 研究证实了传统DLVO理论存在一定局限性. 本研究为高pH环境在胶体迁移中发挥的作用提供了进一步的认识.
胶体;吸附;迁移;饱和多孔介质;pH
近年来,胶体在多孔介质中的迁移行为成为环境领域的热门研究话题.预测胶体在多孔介质中的迁移行为一方面可以评估微生物在地下水中迁移的环境风险[1],另一方面还可以应用于生物修复[2].pH作为影响胶体在饱和多孔介质中迁移的重要因素,学者们已经进行了大量的研究[3-4].由于多孔介质、胶体、微生物表面均携带一定的电荷,由于等电点较低,因此在自然环境下其表面均携带负电荷[5-6],多孔介质与胶体之间存在排斥力.当环境 pH低于某一方的等电点时,表面携带正电荷,则胶体与多孔介质之间存在引力,从而增大了胶体的吸附能力.针对这个吸附量发生变化的临界 pH值,Guan[7]曾研究过病毒在多孔介质中迁移的临界pH值,发现当环境pH值小于微生物或多孔介质的等电点最大值0.5个单位时,病毒的迁移能力会发生极大的改变[7].此外,即使胶体和多孔介质表面均为负电荷的情况下, pH的增加也会导致斥力增大[8].因此许多实验均证实了胶体的吸附能力随着pH的增加而降低[3].然而目前大量的实验对pH的影响研究主要集中于偏中性的环境中,对极酸和极碱环境下 pH影响的研究则较少;此外,已有的研究更倾向于 pH对胶体吸附的影响,而对胶体释放的影响则相对较少.
pH影响胶体迁移过程还有一个重要的原因就是化学异质性.化学异质性指的是由于多孔介质表面存在一些化学杂质,如金属氧化物、有机物等,在自然环境下,这些杂质表面的电荷与多孔介质表面的电荷不同甚至电性相反,产生不同的作用力从而影响了实验结果.化学异质性被认为是造成胶体在多孔介质中的迁移行为与传统的胶体过滤理论(CFT, colloid filtration theory)不符的原因之一[9-10].关于化学异质性学者也进行了大量的研究,理论方面,Duffadar通过计算发现电荷的不均匀会降低排斥势垒从而有利于胶体在初级势阱中的滞留[11],Shen认为当化学杂质大于某一临界值时会提高胶体的不可逆吸附[10];实验方面,Foppen发现石英砂表面的化学杂质有利于提高大肠杆菌的滞留[12],而 Weaver则认为多孔介质表面溶解性有机碳的存在会占据吸附位点从而降低胶体的吸附能力[13].总之,多孔介质表面的化学杂质对胶体迁移的影响是非常显著地.学者通常通过提高溶液的 pH,来消除化学杂质造成的影响[14].通常多孔介质的等电点较低,而部分化学杂质的等电点较高,自然环境下,多孔介质表面带负电荷,而这部分化学杂质表面带正电荷,当提高溶液的 pH后,杂质表面的电荷会由正电荷转变为负电荷,从而降低杂质的影响.
本研究通过比较不同pH环境下胶体的迁移行为,探究了高 pH环境对胶体吸附和解吸行为的影响.此外还结合多孔介质表面化学异质性对胶体吸附的影响,分析讨论了高 pH环境与化学异质性的关系.
1 材料和方法
1.1 胶体和多孔介质
实验中用到胶体为羧基化聚苯乙烯乳胶微球,粒径为 1μm,具有良好的分散性和稳定性,在423nm 波长下用紫外-可见分光光度计来测量.多孔介质为粒径在 0.45mm~0.6mm范围内的玻璃微珠,测得玻璃微珠的孔隙度为0.38.根据玻璃微珠的处理方法我们将其分为两组,一组仅用超纯水漂洗数次,直至上清液的吸光度为 0;另一组先用超纯水漂洗至上清液吸光度为0后,再对其进行酸洗,最后再用超纯水漂洗数十次直至上清液的 pH和电导率均接近超纯水,然后烘干.酸洗方法:先用丙酮和正己烷润洗玻璃微珠,再将玻璃微珠浸泡在浓盐酸中 24h[15].用 SEM-EDS测量两种不同玻璃珠表面的化学组成成分.
1.2 胶体迁移实验
整个实验装置由 4部分组成,包括进液槽、蠕动泵、柱子、部分收集器,装置之间由进液管连接.柱子主体采用 PVC管,中间用湿填法装满玻璃微珠并压实,柱子两侧分别用盖子封口,在玻璃微珠和盖子间铺层孔径为 25μm的尼龙网.液体由进液槽经蠕动泵自下而上流经柱子,最后流入部分收集器中以便检测.所有实验的流速均为2.5mL/min,即达西流速为5.76m/d.整个实验过程需配制不同的背景液和胶体悬液,背景液需在0.01mol/L氯化钠溶液的基础上用氢氧化钠调节pH分别至10、10.5、11、11.5、12,一组不调节pH作为对照组,测得对照组的pH环境为6.8.胶体悬液是在背景液的基础上加入乳胶微球,使浓度达到10mg/L.迁移实验按照玻璃微珠的处理方式分为两组,以采用仅用超纯水漂洗的玻璃微珠作为多孔介质的称为空白组,采用酸处理的玻璃微珠为多孔介质的称之为酸洗组.
胶体迁移实验由胶体吸附实验和胶体解吸实验.在吸附实验中,将酸洗组和空白组分为三个阶段,第一阶段先通入约 20PV的背景液来平衡柱子环境,然后再依次通入 3PV的胶体悬液和5PV的背景液;第二阶段则通入 6PV的超纯水;第三阶段则分别通入pH与背景液相同约11PV的超纯水(也用氢氧化钠调节),对照组则继续通入超纯水.实验期间用部分收集器每隔 5分钟收集流出液,并测量胶体的浓度并绘制穿透曲线.将每一阶段胶体流出液的浓度进行积分,除去总的胶体输入量即得每一阶段的胶体恢复率R1、R2、R3,总的恢复率用RT表示,则RT= R1+ R2+ R3.在胶体解吸实验中,仍然将实验分为空白组和酸洗组.三个阶段和上一实验的步骤方法均相同,唯一的不同是这次的背景液和胶体悬液仅由0.01mol/L氯化钠组成,不调节pH,而在第三阶段依旧加入不同 pH的超纯水.同样地,也计算每一阶段的恢复率R1、R2、R3和总恢复率RT.
1.3 DLVO计算
用Zeta电位分析仪分别测量胶体和两种玻璃珠在不同pH环境下的Zeta电位,计算不同环境下胶体与玻璃珠之间的相互作用能.本次计算采用传统DLVO理论,双电层力采用Hogg等提出的球-平面模型[16],范德华力采用Gregory等提出的球-平面模型[17].
2 结果
2.1 多孔介质的表面化学组成成分
由表 1可以看出,水漂洗后玻璃珠表面多孔介质表面的化学杂质以碳为主,钠和铝和存在一定的量.而经过酸处理后,碳的量由原来的 19.2%减少到 8%,钠和铝的含量也有一定程度的下降,甚至铝的含量已经检测不出来了.这表明酸洗确实可以大大的减小多孔介质的表面化学杂质,但却无法完全的清洗干净[18].

表1 多孔介质表面的化学杂质百分含量(%)Table 1 Surface chemistry composition of porous mediain weight percentage
2.2 Zeta电位与相互作用能
表2列举了在0.01mol/L氯化钠溶液中,不同pH环境下胶体和玻璃珠表面的Zeta电位及相互作用能.对于胶体和玻璃微珠,在自然环境下Zeta电位显负性,随着pH值的增加,Zeta电位逐渐降低导致排斥势垒和次级势阱的升高.而我们通过对比相同pH环境下两种玻璃珠表面的Zeta电位,发现在pH为6.8时,经过酸处理过的玻璃微珠表面的Zeta电位更低,而pH大于等于10时,这时候二者的Zeta电位差并不明显.从计算的相互作用能中可以看出,胶体与玻璃珠之间均存在排斥势垒,表明所有实验环境均为不利环境下的吸附.
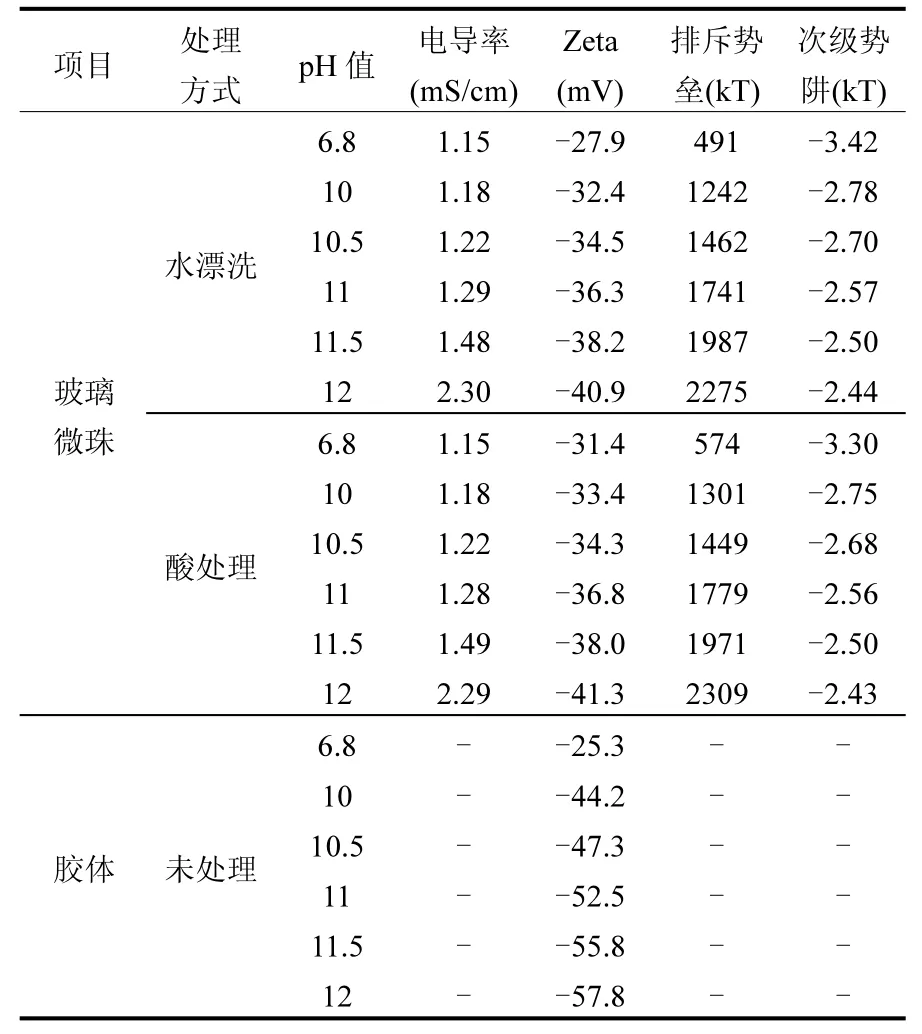
表2 胶体与玻璃珠的Zeta电位以及DLVO相互作用能Table 2 Electro kinetic properties between colloids and glass beads and the calculated DLVO interaction energy
2.3 胶体迁移实验
图 1展示了不同条件下胶体迁移实验流出液中胶体浓度的变化情况.在图c和图d中,由于在前两个阶段每一组均是重复的过程,因而在图像上并没有明显的差异.实验第一阶段,通入胶体悬液后有一定量的胶体被吸附于介质表面,一部分胶体未被吸附从柱子中流出,随着胶体流入量的增加,流出液中胶体浓度迅速增加.当胶体的吸附速率和解吸速率近似平衡后,流出液浓度达到了一个稳定时期(3~5PV).但由于胶体吸附后占据了多孔介质表面的吸附位点,造成吸附速率降低,因此在稳定时期胶体的相对浓度会缓慢上升,称为 Blocking现象[19].尽管存在排斥势垒,但在pH值较低时,仍然有大量的胶体吸附于介质表面,这与传统DLVO理论相矛盾[1].不同pH环境下,胶体和多孔介质之间的斥力不一样,造成流出液浓度也不一样.pH的增加会导致排斥势垒的增加,从而降低胶体的吸附行为,胶体流出液浓度自然也就越高.再次通入背景液后,柱子中残留的胶体和吸附在多孔介质表面的胶体一部分被释放出来,流出液中胶体的浓度急剧下降.实验第二阶段,通入超纯水后会扩大胶体和多孔介质表面的双电层,增加胶体与多孔介质间的排斥力,导致吸附在次级势阱中的胶体解吸下来[20].实验第三阶段,通入高pH的超纯水后,一方面pH升高导致胶体表面的Zeta电位降低,增加了胶体与介质间的排斥力;另一方面 pH升高导致介质中化学杂质表面电性由正电荷转向负电荷,有利吸附转变成不利吸附,使一部分胶体从多孔介质表面解吸出来[4].
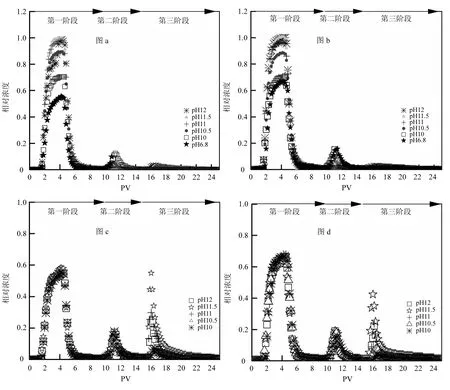
图1 胶体在玻璃珠中的穿透曲线Fig.1 Breakthrough curves of colloids in glass beads
图1中各阶段胶体的恢复率如表3所示.在吸附实验中(图a组和图b组),随着pH的升高,R1也随之增加,并且在 pH达到 11后恢复率接近100%.而R2和R3的恢复率则非常少,也就是说并没有太多的胶体从多孔介质表面解析下来.横向比较空白组和酸洗组,在pH 6.8的环境下二者有明显差异,而在高 pH环境下每一阶段的差别并不明显.也就是说在高 pH环境下,多孔介质表面的化学杂质对胶体的迁移并没有显著的影响.在解吸实验中(图c组与图d组),R1和R2并没有明显的差别,而R3会随着pH的增加而增加(pH为12条件下除外),这也造成了RT和R3一样的规律.若横向比较空白组与酸洗组的差别,酸洗组的R1和RT均要高于同等条件下的空白组,而酸洗组的R3则要略小于空白组.

表3 不同条件下穿透曲线的胶体恢复率Table 3 The recovery of colloids under different conditions
3 讨论
3.1 pH对胶体吸附的影响
由表 1可知,本实验采用的玻璃微珠表面存在一定量化学杂质.酸洗过后可以明显降低这些杂质的含量.去除杂质后会降低多孔介质表面的Zeta电位,从而增大了胶体与多孔介质间的排斥势垒(由表2可知).通过比较在中性pH环境下胶体的迁移行为,可以明显的发现在第一阶段酸洗组胶体的流出液更多.由表3可知空白组在第一阶段的恢复率明显低于酸洗组的恢复率,这说明空白组对胶体的吸附能力更好.在第二阶段,空白组和酸洗组在流出时间和流出浓度上二者相差不大,表明化学杂质对胶体在次级势阱上的吸附并没有显著的影响.因此可以得出结论:化学杂质对胶体的吸附主要是在初级势阱中的吸附.
在吸附实验中,尽管不同 pH环境下的吸附环境均是不利吸附,但 pH对胶体的吸附还是存在明显的影响.当溶液环境的pH为6.8时,胶体流出最少,而随着 pH的增加,胶体恢复率也越来越高.随着 pH进一步提高胶体吸附的能力就越差.当环境pH提高至11,则胶体的恢复率接近100%,即表明胶体几乎不发生吸附行为,此时吸附环境才真正进入了不利环境.DLVO计算结果和真实的实验结果确实存在较大偏差.在第二阶段,当溶液pH越低,则流出液浓度越高,在pH环境为11或更高时,几乎没有胶体流出.这是由于 pH增大会使次级势阱便得更浅(由表 2可知),因此很少有胶体吸附在次级势阱中.
对于pH为6.8的自然环境来说,化学杂质的多与少对胶体的迁移有很大的影响.然而在高pH的环境下,化学杂质对胶体的迁移几乎没有任何影响.当环境pH为10时,空白组和酸洗组的R1分别为76.2%和78.1%,仅仅相差不到2%,而当pH升至10.5时,R1分别达到了95.2%和92.4%,在存在实验误差的结果下,空白组的恢复率反而更高.可以看出,当环境的pH升至10或10以上后,化学杂质对胶体迁移的影响则可以忽略不计.例如,Shen等通过将溶液的pH调至10来消除表面杂质带来的影响[14],而Tufenkji等[21]则将溶液的pH调至11.
3.2 pH对胶体解吸的影响
使胶体从多孔介质表面解吸下来通常有两种办法:提高流速和降低离子强度[22].而本实验中,在不利环境下,通入高 pH的超纯水同样使胶体解吸下来.为了排除离子强度的影响,我们配置了和pH 12超纯水离子强度相同的氯化钠溶液,于第三个阶段通入该氯化钠溶液,在空白组中和酸洗组中均没有发现有胶体解吸下来(数据没有展示).因此实验中胶体的解吸定是由高pH导致的.实验第三阶段,高 pH超纯水可以再次使部分胶体从介质表面解吸下来.并且在同等 pH环境下,空白组中胶体的恢复量均要小于酸洗组,因此这部分胶体释放一部分原因要归功于介质表面的化学杂质.当溶液 pH升高后,导致部分化学杂质的表面电荷由正电荷转向负电荷,有正负相吸转变为负负相斥,造成胶体的解吸.也就是说,化学杂质既可以增加胶体在初级势阱中的吸附,也可以使胶体在高pH环境下解吸更加容易.
本实验中,不管是酸洗组还是空白组,第三阶段恢复率R3随着pH的增加而增加(pH为12除外).pH的增加同样会导致胶体和多孔介质表面的Zeta电位降低,增加胶体和多孔介质间的排斥力(由表2可知).当pH为12时,流出液胶体浓度却有一定程度的下降,这可能和离子强度增加有关.因为离子强度的增加能压缩双电层,增加多孔介质对胶体的吸附能力[23].由于吸附在次级势阱中的胶体已提前被释放出来,并且根据DLVO计算结果表明次级势阱非常浅,仅有几个 kT,不可能吸附大量的胶体,因此这部分胶体是吸附在初级势阱中的.此外,从释放速率来看,在第二阶段释放出来的胶体从最高峰到接近 0处用了大约4PV(11~15PV),而第三阶段释放出来的胶体从最高峰到0点用了大约8PV(16~24PV).有学者将胶体的解吸速率分为快和慢两种[4],而本文中第二阶段和第三阶段胶体的释放速率明显不同,因此来源于不同吸附位点,综上可以判断第三阶段释放的胶体来源于初级势阱.在一些研究中也曾发现胶体从初级势阱中解吸下来[10,24-26].例如,Shen等发现一些因粗糙度而吸附在初级势阱中的胶体会随着溶液离子强度的降低而释放出来[10].根据传统DLVO理论,初级势阱具有无限深的特点,吸附到初级势阱中的胶体不可能被解吸下来.然而胶体和多孔介质间的作用力除了双电层力和范德华力外,还包括水合力、生斥力、溶解力、路易斯酸碱作用力等[27].在传统DLVO理论中加入这些作用力后的理论统称为扩展 DLVO理论.Shen等在探究中考虑了双电层力、范德华力和生斥力后,胶体与多孔介质间的初级势阱不再是无限深的[25],甚至在一些特殊环境下初级势阱呈现正值[26].这些均为胶体从初级势阱中解吸下来提供了可能.而目前的扩展DLVO理论仍然处于起步阶段,还无法对胶体的吸附解吸行为进行很好的解释.
通过比较吸附实验和解吸实验,尽管最终的环境相同,但却有不同量的胶体吸附在多孔介质表面.这里以空白组pH为11为例,在吸附实验和解吸实验中胶体的总恢复率分别达到了103%和75%,解吸实验组仍然有 25%的胶体吸附在介质表面,而吸附实验组几乎没有胶体吸附上去.在pH为 11的环境下,几乎没有胶体可以吸附到介质表面,而对于已经吸附到介质表面的胶体,我们将溶液的pH提高至11,吸附在介质表面的胶体也未必会解吸下来.这表明胶体的吸附不仅和最终的环境有关,也与中间的吸附过程有关.
4 结论
实验结果表明,不利条件下增加溶液的 pH值可以使吸附在初级势阱中的胶体解吸下来,解吸的数量随着 pH值的升高而增加.化学杂质有利于胶体在初级势阱中的吸附,然而因化学杂质而发生吸附的胶体会随着环境pH值的升高而释放出来.而当溶液环境pH为10或高于10的情况下,化学杂质对胶体吸附过程的影响可忽略不计.胶体吸附的多少不仅与中间胶体的吸附解吸过程有关,也和溶液的环境,尤其是高pH环境有关.此外,传统DLVO理论计算结果与实验结果仍然存在一定的偏差,表明了DLVO理论存在局限性.本研究对高pH环境对胶体迁移的影响作了进一步的研究,为将来的研究提供实验上的依据.
[1]Molnar I L. Predicting colloid transport through saturated porousmedia: A critical review [J]. Water Resources Research, 2015, 51(9):6804-6845.
[2]Rogers B. Bacterial transport in NAPL-contaminated porous media [J]. Journal of environmental engineering, 2000,126(7): 657-666.
[3]Minyoung K. Factors that influence the transport of Bacillus cereus spores through sand [J]. Water, Air, & Soil Pollution, 2009,199(1):151-157.
[4]Bradford S A. Equilibrium and kinetic models for colloid release under transient solution chemistry conditions [J]. Journal of Contaminant Hydrology, 2015,181:141.
[5]Foppen J W A. Evaluation of data from the literature on the transport and survival of Escherichia coli and thermotolerant coliforms in aquifers under saturated conditions [J]. Water Research, 2007,40(3):401.
[6]Molnar I L. Impact of surfactant-induced wettability alterations on DNAPL invasion in quartz and iron oxide-coated sand systems [J]. Journal of Contaminant Hydrology, 2010,119(1-4): 1-12.
[7]Guan H. The effect of critical pH on virus fate and transport in saturated porous medium [J]. Groundwater, 2003,41(5):701-708.
[8]Kinoshita T. Bacteria transport in a porous medium: Retention of bacillus and pseudomonas on silica surfaces [J]. Water Research, 1993,27(8):1295-1301.
[9]Bradford S A. Colloid interaction energies for physically and chemically heterogeneous porous media [J]. Langmuir, 2013, 29(11):3668-3676.
[10]Shen C. Influence of surface chemical heterogeneity on attachment and detachment of microparticles [J]. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 2013,433: 14-29.
[11]Duffadar R D. Interaction of micrometer-scale particles with nanotextured surfaces in shear flow [J]. Journal of colloid and interface science, 2007,308(1):20-29.
[12]Foppen J. Transport of E. coli in columns of geochemically heterogeneous sediment [J]. Water Research, 2005,39(13):3082-3088.
[13]Weaver L. Transport of microbial tracers in clean and organically contaminated silica sand in laboratory columns compared with their transport in the field [J]. Science of the Total Environment, 2013,443:55-64.
[14]Shen C. Surface roughness effect on deposition of nano-and micro-sized colloids in saturated columns at different solution ionic strengths [J]. Vadose Zone Journal, 2011,10(3):1071-1081.
[15]Li X. Apparent decreases in colloid deposition rate coefficients with distance of transport under unfavorable deposition conditions: A general phenomenon [J]. Environmental Science & Technology, 2004,38(21):5616-5625.
[16]Hogg R. Mutual coagulation of colloidal dispersions [J]. Transactions of the Faraday Society, 1966,62:1638-1651.
[17]Gregory J. Approximate expressions for retarded van der Waals interaction [J]. Journal of Colloid and Interface Science, 1981, 83(1):138-145.
[18]Johnson W P. Direct observations of colloid retention in granular media in the presence of energy barriers, and implications for inferred mechanisms from indirect observations [J]. Water Research, 2010,44(4):1158-1169.
[19]Bradford S A. Transport and straining of E. coli O157: H7in saturated porous media [J]. Water Resources Research, 2006, 42(12).
[20]Mcdowell-Boyer L M. Particle transport through porous media [J]. Water Resour. Res, 1986,22(13):1901-1921.
[21]Tufenkji N. Breakdown of colloid filtration theory: Role of the secondary energy minimum and surface charge heterogeneities [J]. Langmuir, 2005,21(3):841-852.
[22]Torkzaban S. Colloid release and clogging in porous media: Effects of solution ionic strength and flow velocity [J]. Journal of contaminant hydrology, 2015,181:161-171.
[23]马雪姣.冠状病毒IBV和噬菌体MS2在饱和多孔介质中的运移规律 [J]. 中国环境科学, 2007,27(2):255-259.
[24]Shen C. Heteroaggregation of microparticles with nanoparticles changes the chemical reversibility of the microparticles' attachment to planar surfaces [J]. Journal of Colloid & Interface Science, 2014,421:103-113.
[25]Shen C. Coupled factors influencing detachment of nano-and micro-sized particles from primary minima [J]. Journal of contaminant hydrology, 2012,134:1-11.
[26]Wang Z. Spontaneous Detachment of Colloids from Primary Energy Minima by Brownian Diffusion [J]. Plos One, 2016, 11(1):e0147368.
[27]Bergendahl J. Prediction of colloid detachment in a model porous media: hydrodynamics [J]. Chemical Engineering Science, 2000, 55(9):1523-1532.
Impacts of high pH on transport of colloid in saturated porous media.
YUAN Rui-qiang1*, GUO Wei1,2, WANG Peng3, WANG Shi-qin4(1.School of Environment and Resource, Shanxi University, Taiyuan 030006, China;2.Institute of Resources and Environment Engineering, Shanxi University, Taiyuan 030006, China;3.Key Laboratory of Poyang Lake Wetland and Watershed Research, Ministry of Education, Jiangxi Normal University, Nanchang 330022, China;4.key Laboratory of Agricultural Water Resources Research, Center for Agricultural Resources Research, Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, Shijiazhuang 050021, China). China Environmental Science, 2017,37(9):3392~3398
The adsorption behaviors of colloid transported in untreated glass bead and acid-washed glass bead were compared to examine influences of high pH conditions. Results showed that adsorption of colloid reduced by increasing pH value due to detachment of colloid from the primary minimum under unfavorable conditions. Chemical impurities promoted attachment of colloid to primary minimum. However, a part of colloids attached by chemical impurities was released as pH value increased. Furthermore, the influence of chemical impurities on adsorption of colloid can be ignored when the pH value of solution exceeded 10. The amount of colloid adsorbed depends on not only the adsorption and desorption processes but also the final environment of solution, especially the high pH conditions. The limitation of the traditional DLVO theory was verified. The understanding about the role of high pH conditions during the process of colloid transport was improved.
colloid;adsorption;transport;saturated porous media;pH
X131
A
1000-6923(2017)09-3392-07
2017-01-26
国家自然科学基金项目(41301033)
* 责任作者, 副教授, rqyuan@sxu.edu.cn
袁瑞强(1980-),男,山西太原人,副教授,博士,主要从事流域水循环与水环境研究.发表论文30余篇.
猜你喜欢
数学年刊A辑(中文版)(2022年3期)2023-01-05 10:03:50
牡丹江师范学院学报(自然科学版)(2022年4期)2022-11-21 07:29:38
数学物理学报(2022年4期)2022-08-22 04:07:08
山东冶金(2022年2期)2022-08-08 01:51:22
石材(2022年1期)2022-05-23 12:48:34
数学物理学报(2021年6期)2021-12-21 06:24:02
山西大同大学学报(自然科学版)(2021年4期)2021-08-24 08:38:30
工业设计(2016年11期)2016-04-16 02:48:43
材料科学与工程学报(2016年5期)2016-02-27 07:11:17
大连工业大学学报(2015年4期)2015-12-11 04:06:50
