
儿童MSN基因突变致原发性免疫缺陷病1例并文献复习
2017-09-22 06:29惠晓莹孙金峤王文婕吴冰冰王晓川
中国循证儿科杂志 2017年4期
惠晓莹 孙金峤 王文婕 王 莹 吴冰冰 王晓川
·论著·
儿童MSN基因突变致原发性免疫缺陷病1例并文献复习
惠晓莹 孙金峤 王文婕 王 莹 吴冰冰 王晓川
目的 报告儿童MSN基因突变致原发性免疫缺陷病的临床特征及免疫表型。方法 总结分析1例MSN基因突变致原发性免疫缺陷病患儿的临床资料、免疫表型及治疗,并复习相关文献。结果 患儿男,8岁,临床表现为生后反复呼吸道和消化道感染,反复皮肤湿疹,频发足癣。中性粒细胞、淋巴细胞、单核细胞均降低,免疫球蛋白IgG、IgA和IgM低下,T、B和NK淋巴细胞计数降低,CD4+/CD8+比例倒置,DNT细胞比例增高,基因检测发现MSN基因外显子5有1个半合子、错义突变位点(c.511C>T:p.R171W),为自发突变。在PubMed、Web of Science、中国知网、维普数据库和万方数据库中检索儿童Moesin(MSN)基因突变或缺陷,检索时间均从建库至2017年6月30日。共检索到相关文献2篇,均为英文文献,总结包括本文1例在内的6例MSN基因突变患儿的临床和免疫特点;临床均表现为生命早期发生反复感染,累及呼吸道、消化道和皮肤等,对细菌、真菌和病毒均易感,水痘-带状疱疹病毒感染尤为突出,易累及多系统。免疫表型方面,CD8+T细胞过量表达衰老细胞标志物CD57;给予免疫蛋白替代治疗以及预防性抗生素,可有效减少感染发生。结论MSN基因突变所致免疫缺陷病表现为2岁以内即发生的反复感染,白细胞降低,低丙种球蛋白血症。
MSN基因突变; 淋巴细胞减少; 低丙种球蛋白血症; 原发性免疫缺陷病
1 病例资料
男,8岁。因“反复感染8年余,伴白细胞减少”于2016年12月23日就诊于复旦大学附属儿科医院。
患儿生后3个月开始出现反复呼吸道感染,表现为咳嗽、咳痰、腹泻和发热等,间隔10余天至数月,院外多次血常规提示WBC(1.9~4.8)×109·L-1,中性粒细胞(0.8~3.4)×109·L-1,淋巴细胞(0.7~1.1)×109·L-1。每次抗感染治疗2~5 d可好转。病程中反复出现皮疹,呈湿疹样表现或糠屑样改变。3岁时出现与发热无关的水疱样皮疹。骨髓检查提示感染骨髓象,心脏超声和腹部B超未见异常,为进一步诊治来我院就诊。
患儿父母否认近亲结婚,无家族遗传病史,家族成员无类似疾病史。
体格检查:身高 121 cm;体重 28 kg。精神反应可。左肘部及下肢各有1处糠屑样皮疹,双足底皮肤干燥、脱皮;浅表淋巴结未触及;口腔黏膜光滑,咽不红,双侧扁桃体无肿大;双肺呼吸音粗,未闻及啰音;心音有力,律齐,无杂音;腹软,肝、脾肋下未及;四肢肌力、肌张力正常;神经系统检查未见异常。
一般实验室检查:血常规WBC 1.0×109·L-1,中性粒细胞0.53×109·L-1,淋巴细胞0.4×109·L-1,单核细胞0.03×109·L-1,RBC、Hb和PLT在正常值范围。血涂片未见异型淋巴细胞。降钙素原 0.09 ng·mL-1,尿常规、粪常规、肝肾功能、电解质和凝血功能未见异常,IL-6和血沉在正常值范围,抗-HBs 39.2(0~10)mIu·mL-1,梅毒RPR试验和梅毒螺旋体明胶凝集试验均阴性,HIV抗体阴性。
影像学:胸部CT示右中肺少许渗出。腹部B超示肝、脾、双肾、输尿管和膀胱未见局灶性占位,双侧髋关节、膝关节、踝关节和肘关节未见异常。左手正位骨龄约9岁。
病原学:血CMV-DNA阴性,EBV-DNA和EBV-IgM阴性,外周血白细胞EBV-DNA 2.54×103copies·mL-1,血呼吸道病毒抗体阴性。
免疫功能:T、B、NK淋巴细胞明显减少,初始CD4+T细胞比例降低(21.9%)、中央记忆性CD4+T细胞比例增高(64.9%),转换B细胞比例增高(64.9%),双阴性DNT(TCRαβ+DNT)细胞增多,流式细胞检测图见图1。免疫球蛋白IgG、IgM、IgA和IgE均低下。补体和中性粒细胞呼吸爆发功能均正常。
遗传、血液检查:骨髓穿刺提示骨髓增生极度活跃,粒系核左移。染色体正常。
基因检测:临床诊断“免疫缺陷病”,取得患儿父母知情同意后,抽取患儿及其父母外周血2 mL,行全外显子组检测(WES)。提取基因组DNA(美国Qiagen公司QIAamp DNA Mini试剂盒)后打断、扩增,建立目标基因组库后捕获外显子(美国Illumina公司TruSeqTM外显子富集试剂盒),按照流程(美国Illumina公司Hiseq2500型测序仪)进行PE100测序及测序结果筛选(Q20标准),初步分析数据后进入已建立的WES数据分析流程逐步筛选。检测到符合患儿临床表型及免疫表型的致病基因MSN基因第5号外显子c.511C>T半合子变异,为X染色体隐性遗传,患儿父母该位点无变异,为自发突变。该变异为HGMD数据库已报道的致病性突变。图2显示该突变的Sanger测序验证。

图1 患儿T、B淋巴细胞分型检测流式图
注 A:总T细胞分群:CD3+CD4+T(35%); CD3+CD8+T(54.7%); CD3+CD4-CD8-DNT(7%); B:CD4+T细胞分群:CD3+CD4+D45RA+CD27+初始 T(21.9%),CD3+CD4+CD45RO+CD27+中央记忆T(64.9%),CD3+CD4+CD45RO+CD27-效应记忆 T(11.5%); CD3+CD4+CD45RA+CD27-TEMRA T(1.7%); C:CD8+T细胞分群:CD3+CD8+D45RA+CD27+初始 T(49.1%),CD3+CD8+CD45RO+CD27+中央记忆 T(27.1%), CD3+CD8+CD45RO+CD27-效应记忆 T(5.3%); CD3+CD8+CD45RA+CD27-TEMRA T(18.5%); D:双阴性T细胞分群:TCRαβ+DNT(49.2%); E:记忆B细胞和初始B细胞:CD19+IgD+CD27-初始 B(81.1%),CD19+IgD-CD27+记忆 B(8.1%);F:转换B细胞和浆细胞:CD38++CD24++转换B(64.9%),CD38+CD24-浆细胞(8.1%)
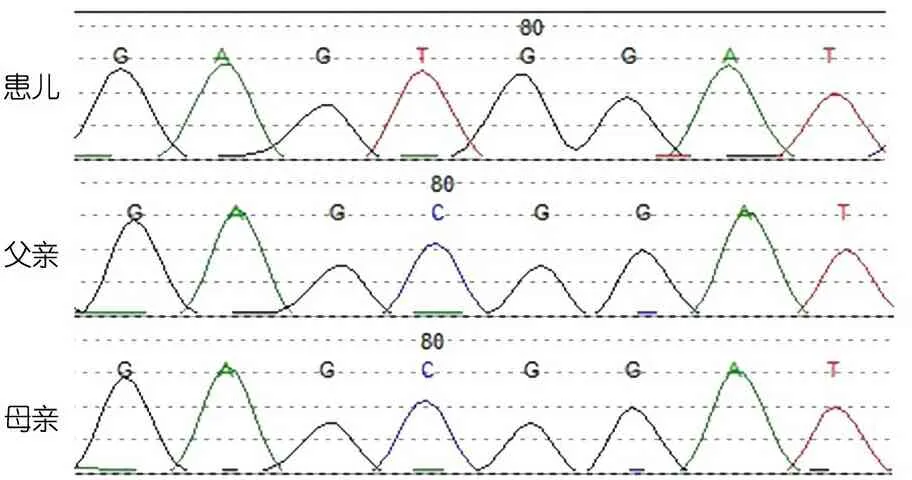
图2 患儿及其父母外周血MSN基因Sanger测序验证
治疗及随访:予罗氏芬抗感染治疗7 d,IVIG(15 g)支持治疗,皮疹予美克外涂,好转出院。出院嘱每月输注IVIG 15 g,复方磺胺甲噁唑口服预防感染,美克外涂皮肤治疗。出院后门诊随访至2017年6月30日,感染情况好转,皮疹好转。
2 复习文献
以“Moesin(MSN) mutation or deficiency”检索Pubmed和Web of Science数据库,以“Moesin(MSN)基因突变或缺陷”检索中国知网、维普和万方数据库,检索时间均从建库至2017年6月30日。共检索到相关文献2篇,均为英文文献,共报告MSN基因突变患儿5例、患者3例,结合本文1例,表1显示6例MSN基因突变患儿的临床特征、免疫功能特点。
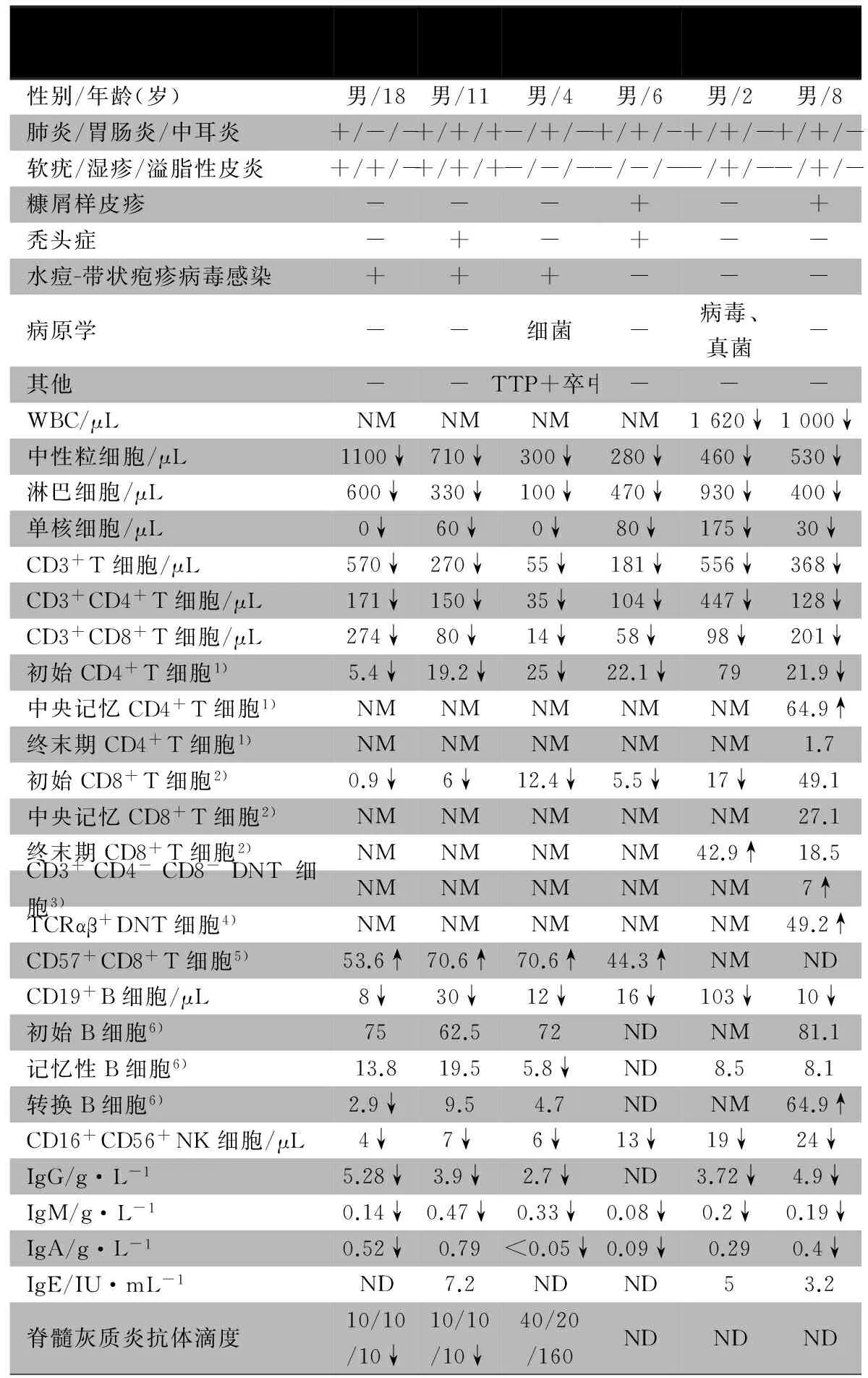
表1 6例MSN基因突变患儿临床特征及免疫功能结果
注 ND:数据未确定;NM:文中未提到;1):占CD4+T细胞百分比;2):占CD8+T细胞百分比;3):占CD3+T细胞百分比;4):占DNT细胞百分比;5):占CD8+T细胞百分比;6):占CD19+B细胞百分比
6例患儿年龄2~18岁,均为男性,来源于不同家庭。①主要临床特点:生后出现反复感染,累及呼吸道、消化道、皮肤,细菌、病毒和真菌均易感,皮肤表现突出、多样,水痘-带状疱疹病毒易呈慢性感染并累及多系统。例3发生血栓性血小板减少性紫癜并发脑卒中,血浆置换后好转。②免疫功能特点:白细胞、淋巴细胞和单核细胞均明显降低,中性粒细胞波动性低下;淋巴细胞各亚类和免疫球蛋白均低下;初始T淋巴细胞比例降低,CD8+T细胞过量表达衰老细胞标志物CD57。
6例患儿MSN均为c.511C>T(R171W)错义突变,其中5例来源于母亲(杂合突变),本文病例为自发突变。
6例患儿的治疗方案为预防性抗生素使用(5例使用复方磺胺甲噁唑)和IVIG,均存活。
3 讨论
Lagresle-Peyrou等[1]在2016年首次提出MSN基因突变使Moesin蛋白表达低下,导致免疫缺陷疾病的发生,MSN基因突变患者表现为生后反复感染,淋巴细胞数目低下、免疫球蛋白降低、淋巴细胞趋化和黏附功能改变等免疫功能缺陷表现,并提出细胞膜结构在维持免疫功能方面有重要作用。
MSN基因定位于Xq11.2~Xq12,属ERM家族成员之一,ERM家族包括Ezrin、Radixin和Moesin3种蛋白。Moesin由3部分构成:靠近细胞膜的N端球形FERM结构域、C端的连接肌动蛋白结构域以及连接N、C两端的α-螺旋中间结构域[2]。ERM蛋白广泛表达于机体细胞中,虽然3种蛋白的结构和功能相似,但不同的器官组织细胞表达的蛋白种类和数目有其差异和特异性。如Moesin在内皮细胞中表达较多,Moesin和Ezrin在造血系统中表达尤为明显[3,4]。Moesin蛋白是细胞膜与细胞骨架的连接蛋白,在维持细胞稳定性方面发挥重要作用,是一些特殊膜结构的组成成员,如微绒毛和伪足等;也是细胞膜相关蛋白和胞质内蛋白信号传导通路的重要部分,影响细胞的移动、变形、黏附以及增殖、分化。Moesin蛋白参与整个免疫系统的活动,主要通过淋巴细胞抗原受体(TCR/BCR)、相关信号蛋白选择、影响淋巴细胞增殖、趋化、黏附等方面发挥重要作用[5-7]。
MSN基因突变患儿表现为早期出现反复多系统感染,对多种病原体均易感,固有免疫和适应性免疫均不同程度受累。患儿对水痘带状疱疹病毒尤为易感,有研究发现[1],MSN突变的淋巴细胞在破伤风类毒素刺激后增殖正常,而在念珠菌属、水痘-带状疱疹病毒刺激后增殖低下,可能机体感染不同病原体后MSN基因对抗感染免疫影响程度不同有关,尚需进一步研究。
文献复习6例MSN基因突变患儿免疫功能检测结果提示,MSN基因突变可同时累及固有免疫和适应性免疫系统,导致免疫细胞数目低下,淋巴细胞发育障碍、功能减低以及凋亡延迟,与Moesin蛋白在免疫系统中的功能研究相一致[5-7]。本例患儿TCRαβ+DNT比例增多,该细胞与自身免疫性疾病相关。有研究[1]报道2例成人MSN基因突变患者(分别42和69岁)病程中未发现有自身免疫性疾病,患儿目前无反复皮疹、关节炎和多脏器损害表现,需密切随访。
Lagresle-Peyrou等[1]发现1例患儿(本文例1)T细胞表达CCR7明显低下,CXCR4部分降低。在CCL21和SDF1a趋化作用下,T细胞的迁移能力明显下降。此外,患儿T细胞对血管细胞黏附分子-1的黏附作用增强,而对纤维连接蛋白的黏附作用正常。可见MSN基因突变影响淋巴细胞趋化和黏附功能,本例患儿中央记忆性CD4+T比例明显增高,可能与淋巴细胞在淋巴器官中迁出和归巢障碍相关。
6例患儿均为c.511C>T(R171W) 位点突变,该位点位于Moesin蛋白N端的FERM区域。有研究[1]证实该突变位点可致淋巴细胞Moesin蛋白表达低下,进而导致淋巴细胞增殖减低和功能异常等一系列免疫功能改变,为致病突变。
对于MSN基因突变患儿,预防性抗生素使用和IVIG可以有效减少感染发生,目前6例患儿生存状况良好,虽然生后反复呼吸道感染,但尚无支气管扩张以及肺功能损害表现,小年龄患儿生长发育正常[1,8]。虽然MSN基因突变患者的免疫表型为累及细胞、体液免疫的联合原发性免疫缺陷,造血干细胞移植可能并非必须的治疗方案。
[1] Lagresle-Peyrou C, Luce S, Ouchani F, et al. X-linked primary immunodeficiency associated with hemizygous mutations in the moesin (MSN) gene. J Allergy Clin Immunol,2016,138(6):1681-1689 .
[2] Sato N, Funayama N, Nagafuchi A, et al.A gene family consisting of ezrin,radixin and moesin.Its specific localization at actin filament/plasma membrane association sites.J Cell Sci,1992,103(1):131-143 .
[3] Shcherbina A, Bretscher A, Kenney DM, et al. Moesin, the major ERM protein of lymphocytes and platelets, differs from ezrin in its insensitivity to calpain. FEBS Lett, 1999,443(1):31-36 .
[4] Berryman M, Franck Z, Bretscher A. Ezrin is concentrated in the apical microvilli of a wide variety of epithelial cells whereas moesin is found primarily in endothelial cells. J Cell Sci,1993,105 ( Pt 4):1025-1043 .
[5] Fehon RG, McClatchey AI, Bretscher A. Organizing the cell cortex cortex:the role of ERM proteins.Nat Rev Mol Cell Biol,2010,11(4):276-287 .
[6] Neisch AL, Fehon RG.Ezrin, radixin and moesin: key regulators of membrane-cortex interactions and signaling.Curr Opin Cell Biol,2011,23(4):377-382 .
[7] Ilani T, Khanna C, Zhou M, et al. Immune synapse formation requires ZAP-70 recruitment by ezrin and CD43 removal by moesin. J Cell Biol. 2007,179(4):733-746 .
[8] Delmonte OM, Biggs CM, Hayward A,et al.First Case of X-Linked Moesin Deficiency Identified After Newborn Screening for SCID. J Clin Immunol. 2017,37(4):336-338
Primary immunodeficiency disease caused by MSN gene mutation in one child and literature review
HUIXiao-ying,SUNJin-qiao,WANGWen-jie,WANGYing,WUBing-bing,WANGXiao-chuan
(ClinicalImmunologyDepartment,Children'sHospitalofFudanUniversity,Shanghai201102,China)
WANG Xiao-chuan, E-mail: xchwang@shmu.edu.cn
ObjectiveTo explore the clinical feature and immunophenotype of primary immunodeficiency disease caused byMSNgene mutation.MethodsClinical data, immunophenotype and treatment in 1 case of primary immunodeficiency disease caused byMSNgene mutation were retrospectively analyzed, and related literatures were reviewed. ResultsAn 8-years-old boy presenting with repeated pulmonary, intestinal infection and recurrent eczema and tinea pedis, was admitted to hospital. Laboratory examination showed that neutrophils, lymphocytes and monocytes were decreased; Immunoglobulin IgG, IgA and IgM were low; T, B and NK lymphocyte counting decreased; the proportion of CD4+/CD8+was reversed and the percentage of DNT cells increased. Whole exome sequencing (WES) was performed and showed a hemi zygote mutation ofMSNgene on the X chromosome (c.511C > T, p.R171W) and was confirmed by Sanger sequencing. The test gene of his parents was normal. The retrieval of "Moesin(MSN) mutation or deficiency" was made in PubMed, web of science, Chinese CNKI, VIP database and wanfang database. From the establishments of these databases to June 30th, 2017, a total of 2 articles were retrieved. Including 1 case of this article, a total of 6 children withMSNmutation were analyzed. All of them showed repeated infection in the early stage of life, involving respiratory tract, digestive tract and skin, and susceptible to bacteria, fungi and viruses. The varicella zoster virus infection was especially prominent and was prone to involve multiple systems. The immunophenotype was similar to that of the case in this article. CD8+T cells overexpressed the senescent marker CD57. The use of immunoglobulin replacement and prophylactic antibiotics was an effective treatment to reduce the incidence of infection.Conclusion The immunodeficiency disease caused byMSNgene mutation is characterized by repeated infection in the early stage of life, decrease of leukocytes and immunoglobulin.
MSNgene mutations; Lymphocytopenia; Hypogammaglobulinemia; Primary immunodeficiency disease
2017-07-31
2017-08-13)
(本文编辑:张崇凡,孙晋枫)
上海市科学技术委员会西医引导项目:14411965400
复旦大学附属儿科医院临床免疫科 上海,201102
王晓川,E-mail:xchwang@shmu.edu.cn
10.3969/j.issn.1673-5501.2017.04.012
猜你喜欢
昆明医科大学学报(2022年4期)2022-05-23
中国医药科学(2022年5期)2022-05-05
中国典型病例大全(2022年7期)2022-04-22
昆明医科大学学报(2021年12期)2021-12-30
临床肝胆病杂志(2021年10期)2021-12-22
天津医科大学学报(2021年4期)2021-08-21
天津医科大学学报(2021年4期)2021-08-21
中国生殖健康(2020年2期)2021-01-18
小学生导刊(2018年13期)2018-06-29
中国生殖健康(2018年2期)2018-01-12
