
肾癌敲基因小鼠模型建立及应用的研究进展
2017-09-20 01:57陈金东
遵义医科大学学报 2017年4期
陈金东
(罗切斯特大学医学中心泌尿科肾癌研究室,纽约罗切斯特市,NY 14642,美国)
候鸟之窗

肾癌敲基因小鼠模型建立及应用的研究进展
陈金东
(罗切斯特大学医学中心泌尿科肾癌研究室,纽约罗切斯特市,NY 14642,美国)
肾癌是一种高度遗传异质性的遗传疾病。目前多个重要的肾癌相关基因已被克隆。这些基因包括VHL、FH、c-Met、TSC1、TSC2、FLCN、PBRM1、BAP1、SETD2 、Trp53、PTEN及APC等。为了研究肾癌的发病机理和测试新的治疗方法,与这些基因相应的敲基因肾癌小鼠模型已经建立。可是,除了TSC1、TSC2、FLCN和Trp53外,大部分单基因敲除小鼠模型未能产生与人类肾癌相似的表型。最近的VHL-PBRM1、VHL-BAP1、PBRM1-BAP1肾特异性双基因敲除小鼠模型普遍产生透明肾细胞癌(CCRCC),是肾癌敲基因小鼠模型发展史上的一个新的里程碑, 为肾癌的研究和药物测试提供了新的工具。肾特异性FLCN敲基因小鼠模型和VHL-Trp53-Kif3a三基因敲除小鼠模型已用于药物试验,并成功地取得了预期的结果。
肾癌;肾癌基因;敲基因小鼠模型; 肾癌治疗
在研究人类疾病发病机理和治疗方法的过程中,由于伦理和安全方面的原因,需要建立疾病动物模型来真实模拟人类疾病,通过这些动物模型探索人类疾病发生和发展机理、发现药物新靶点以及进行临床前药效学评价等。我国在这方面的不足已经影响了相关基础研究和应用基础研究的发展。2017年度国家自然科学基金委员会医学科学部在面上项目中就设立了“疾病动物模型”专项,肯定了人类疾病的动物模型在医学科学研究领域中的理论价值和临床意义。 在所有动物模型中,小鼠基因敲除模型因其可操纵性及其表型的可预期性而被广泛用于基因功能、疾病机制和药物筛选研究。
癌症是当今对人类健康影响最大的一类遗传疾病之一,建立基因敲除小鼠模型来研究癌症的致病机理和探讨其治疗策略就十分有意义。一些癌症已有了较好的敲基因小鼠模型。然而,对于肾癌,基因敲除小鼠模型的研究进程却颇为波折。肾癌主要分为肾透明细胞肾细胞癌(clear cell renal cell carcinoma,CCRCC,80%)、乳头状肾细胞癌(Papillary renal cell carcinoma,PRCC,10%)、嫌色肾细胞癌(Chromophobe renal cell carcinoma,CRCC,5%)、肾集合管癌 (Colleting duct carcinoma) 及尚未分类的肾癌(5%)[1]。已发现的几个重要的肾癌相关基因包括VHL、FH、c-Met、TSC1、TSC2、FLCN、PBRM1、BAP1、SETD2 、Trp53、PTEN及APC等[2]。每个基因被克隆出来后,研究人员就着手建立相应的敲基因肾癌小鼠模型。可是,大部分基因敲除小鼠模型的结果都令人失望,比如其中最重要的VHL、FH、c-Met基因在小鼠肾上敲除后仅偶尔产生肾囊肿外,未见有肾癌产生[3],虽然TSC1/TSC2基因敲除小鼠可以产生肾癌[4],但出现迟、发病率低,不利于药物筛选和药效判断。此后直到FLCN基因的克隆和肾特异性FLCN敲基因小鼠模型的建立及FLCN肾敲基因小鼠能产生多种类型的肾癌,使肾癌基因敲除小鼠模型的研究上了一个新的台阶。最近VHL-PBRM1、VHL-BAP1、PBRM1-BAP1双基因敲除小鼠模型的建立及这些模型能有效地产生与人类相似的透明细胞肾细胞癌,是肾癌研究史上一个新的里程碑。如果从肾癌类型来分,肾癌敲基因小鼠模型可分为透明细胞肾细胞(CCRCC)敲基因小鼠模型、乳头状肾细胞癌(PRCC)敲基因小鼠模型、嫌色肾细胞癌(CRCC)敲基因小鼠模型和综合型肾细胞癌敲基因小鼠模型。本文将分类介绍各种肾癌敲基因小鼠模型的建立及其表型与可能的应用。
1 透明细胞肾细胞癌敲基因小鼠模型
透明细胞肾细胞癌(CCRCC)是一类主要的肾癌,约占肾癌的80%。目前发现与CCRCC相关的基因主要有VHL、PBRM1、BAP1、SETD2。除SETD2外,目前已有VHL、PBRM1、BAP1的单基因敲除小鼠模型和VHL-PBRM1、VHL-BAP1及PBRM1-BAP1的双基因敲除小鼠模型。这些模型中,所有单基因敲除小鼠模型均不能产生肾癌,而所有双基因敲除小鼠模型的表型在程度上既有相似之处,又有一些差别,但均可产生CCRCC。
1.1VHL敲基因小鼠模型VHL基因是第1个克隆到的肾癌抑制基因,是从一种称为 von-Hippel-Lindau的常染色体显性遗传癌症综合症的病人中发现的[5]。VHL病人常因VHL基因突变或基因沉默(gene silencing)而患有肾囊肿和双侧透明细胞肾细胞癌(clear cell renal cell carcinomas,CCRCC)。大部分的CCRCC表现为实体瘤,大约5% 呈现出囊肿样,肿瘤细胞则分布在囊肿壁上。VHL基因位于3p25.3,只有3个外显子,编码大小分别为213个氨基酸和160 个氨基酸的两个蛋白。研究发现大概70%的CCRCC是由VHL突变引起[6-7]。VHL突变在遗传性和散发性的CCRCC都存在。由于80%以上的肾癌属于CCRCC,这一数据表明VHL是一个十分重要的肾癌抑制基因。在VHL克隆后,几个研究小组就着手建立VHL敲基因小鼠模型,因为如果VHL敲基因小鼠能够表现出类似人类的CCRCC,将为肾癌的分子机制和治疗研究提供有力的研究工具。由于传统的敲基因技术会将小鼠全身的VHL基因敲除,致使纯合子VHL敲基因小鼠在胚胎期死亡。为此,Rankin等采用PEPCK(phosphoenolpyruvate carboxykinase promoter) 作为Cre基因的启动子[8],由于PEPCK-Cre只在肾近曲小管和肝细胞中表达,因此VHL-PEPCK-Cre只特异性的敲除肾近曲小管和肝细胞中的VHL基因,这样在一般情况下就不会导致敲基因小鼠在胚胎期死亡。当VHL的外显子1和2 在肾近曲小管中被敲除后,研究者预期敲基因小鼠将产生CCRCC。然而,虽然个别VHL敲基因小鼠能产生肾囊肿或增生(见图1),VHL敲基因小鼠并没有像预期的那样产生肾癌,更没有产生类似人类的透明细胞肾细胞癌[3,8-11]。即使在VHL敲基因小鼠中同时失活Hif基因,也只是引起肾小球囊肿(见图1C)。我们也曾用另一个近曲小管表达启动子Sglt2-Cre来敲除VHL,结果也没有产生肾癌。然后又用Ksp-Cre(Ksp-cadherin gene promoter)作为Cre基因的启动子,来敲除其它肾小管如远曲小管、集合管及亨氏套(loop of Henle),同样未能导致肾肿瘤的发生。这些结果说明以下两点问题:①单一的VHL基因突变尚不能引起肾癌,VHL肾肿瘤可能是多基因突变所致的结果;②小鼠的VHL突变相关的致瘤通路与人类的有一定差异。

A:VHLf/f对照;B:敲基因小鼠产生肾小管囊肿;C:敲基因小鼠在去除Hif基因后产生的肾小囊囊肿。T-Cy,tubular cyst;G-Cy,glomerular cyst。(来自:Cancer Res,2006, 66:2576-2583)图1 VHL敲基因小鼠不产生肾肿瘤
1.2PBRM1敲基因小鼠模型PBRM1是继VHL后发现的第2个重要的肾癌抑制基因,且也位于3号染色体短臂(3p21)[12]。与VHL相似,PBRM1主要与CCRCC有关,有约40% CCRCC是由PBRM1突变引起[12-14],而与其它类型的肾癌几乎没有什么关系[15-16]。PBRM1编码一个1852个氨基酸的BAF180蛋白,而BAF180是SWI/SNF复合物的成员之一,与ATP依赖性的染色体重构有关。当PBRM1被发现也与CCRCC有关时,多个研究组即着手肾特异性PBRM1敲基因小鼠的建立,期望PBRM1基因敲除小鼠模型能产生CCRCC。然而,PBRM1敲基因小鼠与VHL敲基因小鼠一样,不仅没能产生CCRCC,而且也没有产生别的肾癌。我们研究组也采用Ksp-Cre和Sglt2-Cre技术对PBRM1进行了敲除,虽然一部分敲基因小鼠产生肾囊肿,但均未得到预期的肾癌表型。说明PBRM1与VHL一样,在小鼠中单一的PBRM1突变并不能导致CCRCC的产生,CCRCC的产生应是两个以上基因变异的结果。其实研究已经发现,一些肾癌同时有VHL和PBRM1突变[12,17]。而另一研究发现很多转移性肾癌(CCRCC)同时具有BAP1和PBRM1突变[17-18]。这些发现提示CCRCC的产生可能需要同时敲除两个或以上与CCRCC相关的基因。因此,一些研究小组就着手建立起VHL-PBRM1双基因敲除小鼠模型。
1.3VHL-PBRM1双基因敲除小鼠模型 由于VHL和PBRM1的单基因敲除小鼠未能产生预期的肾癌,而一部分透明细胞肾细胞癌同时携带有VHL和PBRM1基因突变[12],这些现象表明,透明细胞肾细胞癌很可能是多基因突变共同作用的结果,如果同时敲除小鼠肾的VHL和PBRM1基因,受累小鼠就很有可能产生CCRCC。为此,Nargund 等采用Ksp-Cre方法敲除了小鼠肾远曲小管、亨氏套管和集合管的VHL和PBRM1基因,获得的VHL-PBRM1双基因敲除模型除了产生肾囊肿外,Nargund等还成功观察到了透明细胞肾细胞癌(见图2)[19]。大概58%的双基因敲除小鼠产生肾囊肿,33%(12/36)的小鼠产生CCRCC。我们小组则采用Sglt2-Cre系统建立了肾近曲小管VHL-PBRM1双基因敲除小鼠模型,同样也观察到了透明细胞肾细胞癌,大约30% (6/21) 的受累小鼠产生肾癌。敲基因小鼠能存活超过12个月以上。这个模型将可用于VHL和PBRM1相关的分子机制的研究,也将能用于药物筛选。这些结果说明小鼠的VHL和PBRM1与人的一样,都与肾癌的发生有关,同时也进一步证明了CCRCC 的产生需要至少两个CCRCC肾癌相关基因的突变才能实现。下面的VHL-BAP1双基因敲除小鼠模型也能说明这一点。
1.4VHL-BAP1双基因敲除小鼠模型 前面已经描述VHL和PBRM1为两个主要的CCRCC 相关基因。后来发现BAP1基因也是一个CCRCC相关基因,BAP1在CCRCC的突变率大约为15%[12,17,20],是第3个最重要的CCRCC相关基因。研究还发现,PBRM1一般与较低级别的CCRCC有关,而BAP1突变则引起高级CCRCC。有趣的是,与VHL和PBRM1一样,BAP1基因也位于人的3号染色体短臂(3p21.1),含有17个外显子,编码一个729个氨基酸的BAP1蛋白,BAP1也叫BRCA1相关蛋白1,是一个去泛素化的泛素分解酶。为了在小鼠中了解BAP1基因突变与肾癌的关系,Wang等采用Six2-Cre系统建立了BAP1肾特异性敲基因小鼠模型[20],发现纯合子BAP1敲基因小鼠在出生后即表现为病态,而且个体比正常对照小鼠要小,大约30 d后死于高血尿氮引起的尿毒症。肾脏多处出现肾囊肿,而且这些囊肿似乎在出生时即已经形成。BAP1杂合子敲基因小鼠却没有肾囊肿等肾损伤发生,更没有观察到CCRCC的出现,因此BAP1单基因敲除小鼠模型也不能产生CCRCC。考虑到VHL也是CCRCC的重要基因,Wang等进一步建立了VHL-BAP1双基因敲除小鼠模型[21]。由于VHL-BAP1纯合子敲基因小鼠(Six2-Cre;VHLlf/f;BAP1f/f)出生后一个月内死亡,只有VHL纯合子BAP1杂合子(Six2-Cre;VHLlf/f;BAP1f/+)小鼠能够存活至少6个月以上,并产生肾囊肿和较小的初级CCRCC。由于BAP1的杂合性,致使所产生的CCRCC不能长大或恶性化。因此,VHL-BAP1肾特异性双基因敲除小鼠与VHL-PBRM1类似,也能产生CCRCC。
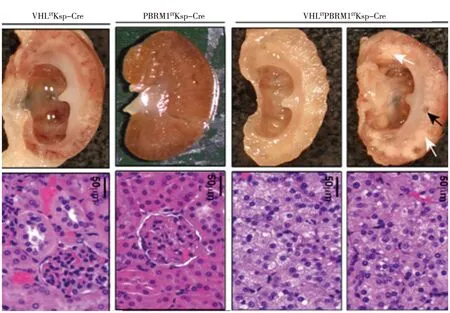
(来自: Cell Reports, 2017,18: 2893-2906)图2 肾特异性VHL-PBRM1双基因敲除小鼠产生CCRCC肾癌
1.5PBRM1-BAP1双基因敲除小鼠模型 虽然VHL、PBRM1和BAP1都位于人的3号染色体短臂上,且均在一个50 MB的区域,但在小鼠中,VHL位于6号染色体上,而PBRM1和BAP1均位于14号染色体上。因此,在小鼠中,PBRM1和BAP1的关系可能更为密切,如果同时敲除小鼠肾中PBRM1和BAP1,被敲除基因的小鼠更易于产生肾肿瘤。PBRM1和BAP1突变都可引起CCRCC,但二者引起的CCRCC 在表型上却有差异。PBRM1突变所致的肾癌为低恶性度CCRCC,而BAP1引起的肾癌为高恶性度CCRCC。患有BAP1突变CCRCC的病人的生存期明显低于患有PBRM1突变CCRCC的病人[22]。最近,Gu等采用Pax8-Cre系统建立了BAP1-PBRM1双基因敲除小鼠模型(见图3)[23],并观察到BAP1-PBRM1双基因敲除小鼠产生高恶性度CCRCC,与人类CCRCC十分相似。这一双基因敲除小鼠模型进一步说明CCRCC的产生需要两个或两个以上相关基因的突变,且PBRM1和BAP1在小鼠中的相互作用比VHL和PBRM1或VHL和BAP1要强。综合分析以上三个双基因敲除小鼠模型的表型,VHL-PBRM1产生恶性度低的CCRCC,而PBRM1-BAP1则可产生恶性度高的CCRCC,二者均能用于CCRCC发病机制的研究,还将可用于抗CCRCC药物的筛选和药效测试。而VHL-BAP1因纯合子敲基因小鼠出生后不久将死亡,杂合子(Six2-Cre;VHLlf/f;BAP1f/+)小鼠表型强度和效率较低,使其应用受到限制。

上一行为恶性度低的肾肿瘤;下一行为恶性度高的肾肿瘤(来自:Cancer Discovery, 2017,DOI: 10.1158/2159-8290.CD-17-0292)图3 肾特异性PBRM1-BAP1双基因敲除小鼠产生CCRCC肾癌
2 乳头状肾细胞癌小鼠模型
前面已经介绍了CCRCC的肾特异性基因敲除小鼠模型,这里介绍乳头状肾细胞癌(PRCC)基因敲除小鼠模型。PRCC是第二类重要的肾癌,约占所有肾癌的15%,乳头状肾细胞癌又分为I型和II型[24]。目前发现与乳头状肾细胞癌有关的基因有c-Met和FH。
2.1c-Met基因突变小鼠模型c-Met基因是一个原癌基因,位于染色体7q31.2[25]。c-Met编码肝细胞生长因子 (hepatocyte growth factor,HGF) 的一个长为1390氨基酸的受体Met。基因突变导致Met基因激活,致使Met蛋白高度表达,血管生成,导致肿瘤生长及肿瘤转移。在肾脏中,c-Met基因的激活主要与散发的 I 型乳头状肾细胞癌有关,占所有乳头状肾细胞癌的5%左右[26]。为了研究c-Met突变在体内的致瘤性,Graveel 等建立了5种c-Met突变的小鼠模型:D1226N、Y1228C、M1248T、 M1248T 和 L1193V。结果发现D1226N、Y1228C、M1248T 和 L1193V突变小鼠产生大量肉瘤(sarcoma)和淋巴瘤(lymphoma),而M1248T则产生上皮细胞癌(carcinoma)和淋巴瘤。然而,这些c-Met突变小鼠并没有产生类似人类的肾肿瘤,即乳头状肾细胞瘤。不过发现上述肿瘤细胞中存在有6号染色体三体和c-Met突变重复,这一点和人乳头状肾细胞癌的染色体变异相似。
2.2FH敲基因小鼠模型FH是在2002年从遗传性平滑肌瘤病伴乳头状肾细胞癌(Hereditary Leiomyomatosis and Renal Cell Cancer, HRLCC)病人中发现的[27],因此,FH只与人乳头状肾细胞癌(PCRR)有关。FH位于人染色体1q43,编码一个由468个氨基酸组成的线粒体三羧酸循环中延胡索酸水化酶(Fumarate hydratase,FH)。FH主要和 II 型乳头状肾细胞癌有关,而在其它类型的肾癌中并未发现有FH突变。而且这类乳头状肾细胞癌需伴有平滑肌瘤病,也就是FH突变只存在于HLRCC综合症中。为了更进一步研究FH的功能,Pollard等于2007年建立了一个FH敲基因小鼠模型,FH的外显子2和3被敲除。由于FH全身敲基因小鼠在胚胎期死亡,因而又采用Ksp-Cre技术建立了FH肾特异性敲基因小鼠模型,可是FH敲基因小鼠也只产生肾囊肿,未见有肾癌发生(见图4)。由于Ksp-Cre只在远曲小管、集合管和亨氏套中表达,而近曲小管的FH并没有被敲除,是否敲除近曲小管里的FH可以产生肾癌尚不清楚。从现有结果看,与VHL情况类似,仅仅敲除FH基因不足以导致PRCC的生成,还有哪些基因也参与了FH-相关的乳头状细胞癌的产生还有待进一步研究。目前还没有产生PRCC的双基因敲除小鼠模型。

A:FH敲基因小鼠肾脏;B:野生型小鼠肾H&E染片;C:FH敲基因小鼠肾囊肿H&E染片。(来自: Cancer Cell,2007,11,311-319)。图4 FH肾特异性敲基因小鼠模型产生肾囊肿
3 嫌色肾细胞癌敲基因小鼠模型——FLCN敲基因小鼠模型
FLCN是目前唯一一个主要与人类嫌色肾细胞癌(CRCC)有关的基因,但并非是CRCC特异性的,FLCN也与其它类型的肾癌有关。FLCN基因于2002年从Birt-Hogg-Dube综合症 (Birt-Hogg-Dube syndrome,BHD) 病人中克隆获得[28-29]。BHD病人易发皮肤肿瘤,肾癌,肺部囊肿,气胸,及其它症状如肠道息肉。大概50%的BHD家族有肾癌历史[30],有34%的BHD病人有肾癌。人的FLCN位于染色体17p11.2,共有 14个外显子,编码一个579氨基酸的FLCN蛋白,并可能调节mTOR和TGF-β等信号传导途径。FLCN与VHL基因不同,VHL基因突变一般只导致CCRCC的产生,而FLCN突变几乎引起各类肾癌的发生,一项对来自30个BHD家系中的130例肾癌患者的研究发现,FLCN突变出现在34%嫌色肾细胞癌,5%的大嗜酸粒细胞瘤(oncocytoma),50%的嫌色肾细胞癌与大嗜酸粒细胞瘤混合型肾癌,9%的透明细胞肾细胞癌及2%的乳头状肾细胞癌中[31]。FLCN在肾癌中常发生突变,其中以11外显子突变最多[32],其类型有大片段缺失[33]、杂合性丢失[34]、点突变等[35]。此外,在狗和大鼠的肾癌中也发现有FLCN突变[36-38]。与其它肾癌基因不同的还有,FLCN敲基因小鼠模型能复制出人类BHD肾癌。因此,这是目前唯一能同时与各类肾细胞癌有关的基因。
最初建立的FLCN全身敲除小鼠模型的纯合子小鼠在胚胎期就死亡,而杂合子小鼠发病很迟,发病率偏低且不稳定[39-41]。基于上述问题,研究人员又采用Ksp-Cre系统建立了FLCN肾远曲小管-亨氏套管-集合管特异性基因敲除小鼠和FLCN肾近曲小管基因敲除小鼠模型,首次成功建成了具有肾癌表型的肾小管特异的FLCN基因敲除小鼠模型[42-44]。这两个模型中,前者小鼠在出生后一周即出现肾囊肿和细胞增生,随后伴有囊肿腺癌的产生(见图5),小鼠血尿素氮快速增高,三周左右即死于尿毒症。由于受累小鼠生存时间太短,因此并没有实体瘤产生。
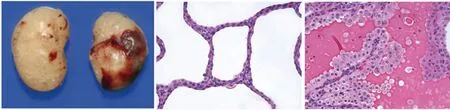
(来自: 本人的研究工作)。图5 FLCN肾远曲小管-亨氏套管-集合管特异性基因敲除小鼠产生肾囊肿和腺癌
与前者不同,肾近曲小管FLCN基因敲出模型能产生各类肾癌(肾囊肿发病率为100%,肿瘤发病率约70%)(见图 6),而且肾癌出现在出生后6~7个月,生存时间长达24个月,因而具有很高的研究应用价值。尤其重要的是,从该小鼠模型能动态观察肾癌的发生发展过程。即在1~2个月时,可观察到肾囊肿;3~4个月时,囊肿增多,可见肾细胞增生;5~6个月时,增生组织向癌转化;6~7个月时,已能观察到微型肾细胞癌,主要为肾嫌色细胞癌;到了10个月左右时,已能观察到中等大小的癌;12个月后,大型肿瘤出现,此时乳头状肾细胞癌开始占主要;16个月后,一些肿瘤已转化为肉瘤状肾癌,一类恶性化程度较高的肾癌[43],很适合做药物筛选和药效研究。我们利用FLCN肾特异性敲基因小鼠的这一特点,用雷帕霉素对小鼠进行了10个月的治疗[43]。由于研究已经证明FLCN缺失导致mTOR信号传导通路的激活,从而促进肾癌细胞的生长,而雷帕霉素是mTOR的抑制剂。雷帕霉素治疗能抑制肾癌的生长,但不能完全消除癌细胞。这说明,FLCN相关癌细胞的产生需要不止一个信号通路的激活。这些结果证明,FLCN肾近曲小管基因敲除小鼠模型可以用于肾癌的分子机制研究和药物的测试筛选。是目前一个较为理想的肾癌敲基因小鼠模型。

(来自: 本人的研究工作)。图6 肾近曲小管FLCN基因敲出模型能产生各类肾癌
4 综合型肾细胞敲基因小鼠模型
综合型肾细胞癌敲基因小鼠模型是指小鼠在基因敲除后可产生多类肾细胞癌,包括CCRCC、PRCC、CRCC、肾集合管癌及其它肾细胞癌。其实前面描述的FLCN敲基因小鼠模型也属于这一类,只是FLCN突变主要与人嫌色肾细胞癌有关,约占50%,就将其视为嫌色肾细胞癌基因敲除小鼠模型。此外,TSC1、TSC2和NF2的敲基因小鼠也产生多类肾细胞癌,故也属于综合型肾细胞癌敲基因小鼠模型。
4.1TSC1/TSC2敲基因小鼠模型TSC1/TSC2是两个紧密相关的基因,均从结节性硬化症(Tuberous sclerosis,TS)病人中发现的[45-46]。由于结节性硬化症病人常有肾癌发生[47],因此也将TSC1/TSC2作为肾癌相关基因,但在散发性肾癌中很少发现有这两个基因的突变。TSC1基因位于染色体9q34.13,而TSC2则位于染色体16p13.3。TSC1基因有23个外显子,编码一个1164个氨基酸的错构瘤蛋白(hamartin),而TSC2基因有41个外显子,编码一个长达1807氨基酸的薯球蛋白(tuberin),两蛋白结合形成蛋白复合物,抑制mTOR信号传导途径。因此,我们一般将这两个基因一起讨论。Kobayashi等于2001年通过传统的基因敲除法去除了TSC1基因的第6、7和8三个外显子,TSC1纯合子敲基因小鼠在胚胎期致死,杂合子能成活至成年,除了产生肾囊肿外,还可产生多种肾囊肿腺瘤,但肾囊肿腺瘤出现的很晚。此外,还发现有肝脏血管瘤和子宫瘤等。TSC2敲基因小鼠的表型与TSC1敲基因小鼠的相似。其实TSC2敲基因小鼠模型早于TSC1敲基因小鼠模型建成。Kobayashi等在敲基因小鼠模型中去除了TSC2基因的第3号和第4号外显子。TSC2纯合子敲基因小鼠也是胚胎期致死,杂合子小鼠也产生肾囊肿和多种肾囊肿腺癌及肝脏血管瘤,但二者的肿瘤出现很晚,发病率很低。因此,这两个模型均不能用于肾癌药物的筛选和药效评价。但可以用于TSC1/TSC2的分子机制的研究。
4.2NF2敲基因小鼠模型NF2基因突变与II型神经纤维瘤(neurofibromatosis)和施万瘤病(schwannomatosis) 的发生有关。NF2有19个外显子,位于染色体22q2.2,编码一个595个氨基酸的神经纤维瘤蛋白(neurofibromin 2或merlin)。由于NF2调节EGFR的表达,而高表达的EGFR在肾癌中很常见,因此,NF2的突变也与肾癌的发生有关。不过NF2基因在肾癌中的突变率只有大约2%。为了进一步了解NF2在肾癌中的作用,Morris于2009年采用了Villin-Cre方法建立了肾近曲小管特异性NF2敲基因小鼠模型[48]。在这个模型中,NF2基因的第2和3号外显子被敲除。几乎所有NF2肾近曲小管纯合子敲基因小鼠在15 d大时就可观察到肾细胞增生和肾囊肿,在3个月大时就长出小的肾小管内肿瘤,当受累小鼠长到6~10个月大时,肾肿瘤已转变为侵害性肾癌,同时也发现这些肾癌中EGFR高度表达,表明NF2的失活导致了EGFR信号通路的激活,从而引起了肾癌的产生。NF2敲基因小鼠的生存期达10个月以上。此外,未发现有杂合子小鼠产生肾癌。虽然NF2不是一个主要的肾癌相关基因,但这个肾癌小鼠模型似乎是一个外显率比较高,发病较早的肾癌模型,理论上应该能用于NF2相关的肾癌发病机制的研究及相关药物的筛选。可惜作者没有进一步利用这个模型进行药物筛选试验。
4.3 其它双基因或三基因肾癌敲除小鼠模型 除了上述双基因敲除小鼠模型外,还有肾VHL-Trp53双基因敲除小鼠模型和肾VHL-PTEN双基因敲除小鼠模型[9-10]。虽然VHL-Trp53敲除小鼠模型也能产生少量体积较小的肾癌[10],但考虑到Trp53是一个广谱抑癌基因,其突变与多种癌症的产生有关, 已经有报道表明Trp53单基因敲除小鼠模型也能导致肾癌的产生,而作者并没有对VHL-Trp53 双基因敲除小鼠模型与Trp53单基因敲除小鼠模型的表型做比较。因此, 很难确定VHL的突变在VHL-Trp53 基因敲除小鼠模型中的真实作用。PTEN是另一个广谱抑癌基因,但肾特异性PTEN基因敲除小鼠模型并没有产生肾细胞癌,而是产生了肾盂泌尿上皮细胞癌(urothelial carcinoma of the renal pelvis)。VHL-PTEN肾双基因敲除小鼠模型除了有肾囊肿外,同样也未能长出肾细胞癌[9]。最近也有研究者建立了肾特异性的VHL-Trp53-Kif3a和VHL-Trp53-Rb1三基因敲除小鼠模型[49-50],虽然这些小鼠也能产生肾癌,但VHL-Kif3a敲基因小鼠只能产生肾囊肿,说明p53 在肾癌的生成中起着决定性的作用。因此,从这些结果看,VHL和p53 或PTEN在功能上并没有明确的互补作用,因此,VHL在这些双基因或三基因的敲除小鼠中的致癌作用并不明显。其应用还需进一步的研究。
5 肾癌小鼠模型的应用前景
随着分子生物学和分子遗传学技术的不断发展,生物信息学的应用以及大数据的出现,人们已对各类疾病发病的分子基础有了更深入的了解和认识。然而对各类遗传疾病的治疗方面的研究却并没有多大进展。其原因之一是没有合适的动物模型来用于各种药物及治疗方案的测试。癌症作为一类复杂的遗传疾病,更需要动物模型作为研究的平台。小鼠因其个体小,繁殖快,生命周期短,其基因组已被全部测序,且95%以上的基因与人类基因相似,是目前相对理想的动物模型研究对象。就癌症而言,癌症小鼠模型体内的肿瘤可以反应人类癌症相关基因和肿瘤的分子及细胞特性,因此,在理论上,癌症的致病机制的研究和新药物的测试及新疗法的检测都可以依赖于经遗传工程精确改造的癌症小鼠模型。然而,几十年来,各类癌症小鼠模型不断出现,但真正有研究及应用价值的癌症小鼠模型却寥寥无几。一个癌症小鼠模型需满足以下四点才可以考虑用于药物测试和新疗法的检验:①产生的癌症和人类的相似;②小鼠癌症发病要早,在一岁以前发病较好;③发病率要高;④发病小鼠的寿命要足够长,以便于有足够的时间观察疗效。以本文提及的肾癌小鼠模型为例,VHL、FH、c-Met、PBRM1、BAP1等肾敲基因小鼠模型均未能产生肾肿瘤,无法用于进一步的肿瘤分子机制及药物测试等研究;而一小部分TSC1、TSC2肾敲基因杂合子小鼠虽能产生肾肿瘤,但肿瘤出现在小鼠生命的晚期,也没法用于药物试验。直到最近出现的肾近曲小管FLCN敲基因小鼠模型和VHL-PBRM1、VHL-BAP1及PBRM1-BAP1等肾双基因敲除小鼠模型的出现,为肾癌的分子发病机制研究及药物测试研究提供了新的希望。目前,肾近曲小管FLCN敲基因小鼠模型已用于雷帕霉素的药物试验,并取得了一定疗效[43],雷帕霉素抑制肾囊肿及肿瘤细胞的生长。因此,肾近曲小管FLCN敲基因小鼠模型将可以用于FLCN相关信号传导通路的研究及更多药物的测试,可为将来的药物临床试验提供有用的实验依据,是一个很有研究价值的肾基因敲除小鼠模型。最近,Harlander 等也利用其建立的VHL-Trp53-Rb1三基因敲除小鼠模型进行了药物试验[50],他们首先采用舒尼替尼(sunitinib) 对4只受累小鼠进行了初级治疗,发现受治疗的19个肿瘤中,3个出现了消退,10 个表现为停止生长或生长很慢,6个继续生长。接着他们又对这四只小鼠用依维莫司(everolimus)进行了二级治疗,发现78% 的肿瘤停止生长或发生消退,只有4个肿瘤产生了抗性。舒尼替尼和依维莫司是临床上已用于肾癌治疗的药物[51]。说明这个模型同样可以用于临床前药物的筛选和新疗法的测试。VHL-PBRM1、VHL-BAP1及PBRM1-BAP1等肾双基因敲除小鼠模型目前还没有用于药物试验[18,21,23],但因这些模型能产生与人类CCRCC相似的肾癌,且发病较早,发病率较高,小鼠生存期较长等特点,也将是很有潜力的肾癌分子机制和药物测试的研究工具。这些模型的应用将会大大加速肾癌药物的研究与开发的速度。
[1] Crumley S M,Divatia M,Truong L,et al.Renal cell carcinoma: Evolving and emerging subtypes [J].World Journal of Clinical Cases,2013,1(9):262-275.
[2]陈金东.肾癌遗传学研究进展 [J].遵义医学院学报,2014,37(2):132-137.
[3]Kleymenova E,Everitt J I,Pluta L,et al.Susceptibility to vascular neoplasms but no increased susceptibility to renal carcinogenesis in vhl knockout mice [J].Carcinogenesis,2004,25(3):309-315.
[4]Qian C N,Knol J,Igarashi P,et al.Cystic renal neoplasia following conditional inactivation of apc in mouse renal tubular epithelium [J].The Journal of Biological Chemistry,2005,280(5): 3938-3945.
[5]Latif F,Tory K,Gnarra J,et al.Identification of the von hippel-lindau disease tumor suppressor gene [J].Science,1993,260(5112):1317-1320.
[6]Linehan W M,Lerman M I,Zbar B.Identification of the von hippel-lindau (vhl) gene.Its role in renal cancer [J].Jama,1995,273(7):564-570.
[7]Zbar B.Von hippel-lindau disease and sporadic renal cell carcinoma [J].Cancer surveys,1995,25(4):219-232.
[8]Rankin E B,Tomaszewski J E,Haase V H.Renal cyst development in mice with conditional inactivation of the von hippel-lindau tumor suppressor [J].Cancer research,2006,66(5):2576-2583.
[9]Frew I J,Thoma C R,Georgiev S,et al.Pvhl and pten tumour suppressor proteins cooperatively suppress kidney cyst formation [J].The EMBO Journal,2008,27(12):1747-1757.
[10]Albers J,Rajski M,Schonenberger D,et al.Combined mutation of vhl and trp53 causes renal cysts and tumours in mice [J].EMBO Molecular Medicine,2013,5(6):949-964.
[11]Pritchett T L,Bader H L,Henderson J,et al.Conditional inactivation of the mouse von hippel-lindau tumor suppressor gene results in wide-spread hyperplastic,inflammatory and fibrotic lesions in the kidney [J].Oncogene,2015,34(20):2631-2639.
[12]Varela I,Tarpey P,Raine K,et al.Exome sequencing identifies frequent mutation of the swi/snf complex gene pbrm1 in renal carcinoma [J].Nature,2011,469(7331):539-542.
[13]Xu X,Hou Y,Yin X,et al.Single-cell exome sequencing reveals single-nucleotide mutation characteristics of a kidney tumor [J].Cell,2012,148(5):886-895.
[14]Pawlowski R,Muhl S M,Sulser T,et al.Loss of pbrm1 expression is associated with renal cell carcinoma progression [J].International Journal of Cancer Journal International Du Cancer,2013,132(2):E11-17.
[15]Joseph R W,Kapur P,Serie D J,et al.Clear cell renal cell carcinoma subtypes identified by bap1 and pbrm1 expression [J].The Journal of Urology,2016,195(1):180-187.
[16]Ho T H,Kapur P,Joseph R W,et al.Loss of pbrm1 and bap1 expression is less common in non-clear cell renal cell carcinoma than in clear cell renal cell carcinoma [J].Urologic Oncology,2015,33(1):23 e29-23,e14.
[17]Gossage L,Murtaza M,Slatter A F,et al.Clinical and pathological impact of vhl,pbrm1,bap1,setd2,kdm6a,and jarid1c in clear cell renal cell carcinoma [J].Genes,Chromosomes & Cancer,2014,53(1):38-51.
[18]Eckel-Passow J E,Serie D J,Cheville J C,et al.Bap1 and pbrm1 in metastatic clear cell renal cell carcinoma: Tumor heterogeneity and concordance with paired primary tumor [J].BMC Urology,2017,17(1):19.
[19]Nargund A M,Pham C G,Dong Y,et al.The swi/snf protein pbrm1 restrains vhl-loss-driven clear cell renal cell carcinoma [J].Cell reports,2017,18(12):2893-2906.
[20]Wang S S,Gu Y F,Wolff N,et al.Bap1 is essential for kidney function and cooperates with vhl in renal tumorigenesis [J].Proceedings of the National Academy of Sciences of the United States of America,2014,111(46):16538-16543.
[21]Gao W,Li W,Xiao T,et al.Inactivation of the pbrm1 tumor suppressor gene amplifies the hif-response in vhl-/- clear cell renal carcinoma [J].Proceedings of the National Academy of Sciences of the United States of America,2017,114(5):1027-1032.
[22]Kapur P,Pena-Llopis S,Christie A,et al.Effects on survival of bap1 and pbrm1 mutations in sporadic clear-cell renal-cell carcinoma: A retrospective analysis with independent validation [J].The Lancet Oncology,2013,14(2):159-167.
[23]Gu Y F,Cohn S,Christie A,et al.Modeling renal cell carcinoma in mice: Bap1 and pbrm1 inactivation drive tumor grade [J].Cancer discovery,2017.
[24]Twardowski P W,Mack P C,Lara P N.Papillary renal cell carcinoma: Current progress and future directions [J].Clinical genitourinary cancer,2014,12(2):74-79.
[25]Cooper C S,Park M,Blair D G,et al.Molecular cloning of a new transforming gene from a chemically transformed human cell line [J].Nature,1984,311(5981):29-33.
[26]Baldewijns M M,van Vlodrop I J,Schouten L J,et al.Genetics and epigenetics of renal cell cancer [J].Biochimica et Biophysica Acta,2008,1785(2):133-155.
[27]Tomlinson I P,Alam N A,Rowan A J,et al.Germline mutations in fh predispose to dominantly inherited uterine fibroids,skin leiomyomata and papillary renal cell cancer [J].Nature genetics,2002,30(4):406-410.
[28]Nickerson M L,Warren M B ,Toro J R,et al.Mutations in a novel gene lead to kidney tumors,lung wall defects,and benign tumors of the hair follicle in patients with the birt-hogg-dube syndrome [J].Cancer cell,2002,2(2):157-164.
[29]Birt A R,Hogg G R,Dube W J.Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons [J].Archives of Dermatology,1977,113(12):1674-1677.
[30]Toro J R,Wei M H,Glenn G M,et al.Bhd mutations,clinical and molecular genetic investigations of birt-hogg-dube syndrome: A new series of 50 families and a review of published reports [J].Journal of Medical Genetics,2008,45(6):321-331.
[31]Schmidt L S,Nickerson M L,Warren M B,et al.Germline bhd-mutation spectrum and phenotype analysis of a large cohort of families with birt-hogg-dube syndrome [J].American Journal of Human Genetics,2005,76(6):1023-1033.
[32]曹磊,邹卫,易龙.Flcn基因与原发性自发性气胸的研究进展 [J].东南大学学报:医学版,2012,31(6):771-776.
[33]Naf E,Laubscher D,Hopfer H,et al.Birt-hogg-dube syndrome: Novel flcn frameshift deletion in daughter and father with renal cell carcinomas [J].Familial Cancer,2016,15(1):127-132.
[34]Schmidt L S,Linehan W M.Molecular genetics and clinical features of birt-hogg-dube syndrome [J].Nature Reviews Urology,2015,12(10):558-569.
[35]Sattler E C,Lang M U,Van Steensel M A,et al.Late onset of skin manifestations in birt-hogg-dube syndrome with flcn mutation p.W260x [J].Acta dermato-venereologica,2012,92(2): 187-188.
[36]Okimoto K,Kouchi M,Matsumoto I,et al.Natural history of the nihon rat model of bhd [J].Current Molecular Medicine,2004,4(8):887-893.
[37]Okimoto K,Sakurai J,Kobayashi T,et al.A germ-line insertion in the birt-hogg-dube (bhd) gene gives rise to the nihon rat model of inherited renal cancer [J].Proceedings of the National Academy of Sciences of the United States of America,2004,101(7):2023-2027.
[38]Lingaas F,Comstock K E,Kirkness E F,et al.A mutation in the canine bhd gene is associated with hereditary multifocal renal cystadenocarcinoma and nodular dermatofibrosis in the german shepherd dog [J].Human Molecular Genetics,2003,12(23):3043-3053.
[39]Hartman T R,Nicolas E,Klein-Szanto A,et al.The role of the birt-hogg-dube protein in mtor activation and renal tumorigenesis [J].Oncogene,2009,28(13):1594-1604.
[40]Hasumi Y,Baba M,Ajima R,et al.Homozygous loss of bhd causes early embryonic lethality and kidney tumor development with activation of mtorc1 and mtorc2 [J].Proceedings of the National Academy of Sciences of the United States of America,2009,106(44):18722-18727.
[41]Hudon V,Sabourin S,Dydensborg A B,et al.Renal tumour suppressor function of the birt-hogg-dube syndrome gene product folliculin [J].Journal of Medical Genetics,2010,47(3):182-189.
[42]Chen J,Futami K,Petillo D,et al.Deficiency of flcn in mouse kidney led to development of polycystic kidneys and renal neoplasia [J].Plos One,2008,3(10):e3581.
[43]Chen J,Huang D,Rubera I,et al.Disruption of tubular flcn expression as a mouse model for renal tumor induction [J].Kidney International,2015,88(5):1057-1069.
[44]Wu M,Si S,Li Y,et al.Flcn-deficient renal cells are tumorigenic and sensitive to mtor suppression [J].Oncotarget,2015,6(32):32761-32773.
[45]Van Slegtenhorst M,De Hoogt R,Hermans C,et al.Identification of the tuberous sclerosis gene tsc1 on chromosome 9q34 [J].Science,1997,277(5327):805-808.
[46]Identification and characterization of the tuberous sclerosis gene on chromosome 16 [J].Cell,1993,75(7):1305-1315.
[47]Borkowska J,Schwartz R A,Kotulska K,et al.Tuberous sclerosis complex: Tumors and tumorigenesis [J].International Journal of Dermatology,2011,50(1):13-20.
[48]Morris Z S,McClatchey A I.Aberrant epithelial morphology and persistent epidermal growth factor receptor signaling in a mouse model of renal carcinoma [J].Proceedings of the National Academy of Sciences of the United States of America,2009,106(24):9767-9772.
[49]Guinot A,Lehmann H,Wild P J,et al.Combined deletion of vhl,trp53 and kif3a causes cystic and neoplastic renal lesions [J].The Journal of Pathology,2016,239(3):365-373.
[50]Harlander S,Schonenberger D,Toussaint N C,et al.Combined mutation in vhl,trp53 and rb1 causes clear cell renal cell carcinoma in mice [J].Nature Medicine,2017.
[51]陈金东.转移性肾癌的临床治疗研究进展 [J].遵义医学院学报,2015,38(3):201-208.
(编辑:谭秀荣)
Advances in development and application of knockout mouse models of kidney cancer
Chen Jindong
(Kidney Cancer laboratory,Department of Urology,University of Rochester Medical Center,Rochester,NY 14642,USA)
Kidney cancer is a highly genetic heterogenic disease.To date,many key kidney cancer-related genes have been cloned.These genes includeVHL,FH,c-Met,TSC1,TSC2,FLCN,PBRM1,BAP1,SETD2,PTEN,Trp53,andAPC.To investigate renal pathogenic mechanisms and test new therapeutic strategies,corresponding knockout kidney cancer mouse models have been developed following the identification of these genes.However,most of the single-gene knockout models failed to recapitulate the human renal cancer except fromTSC1,TSC2,FLCN,andTrp53 models.The development ofVHL-PBRM1,VHL-BAP1,andPBRM1-BAP1double-gene knockout mouse models has represented a new milestone in kidney cancer research and provided new tools for the investigation of kidney cancer and drug test.Kidney-specificFLCNandVHL-Trp53-Kif3atriple-gene knockout mice have been used for renal cancer drug test, which successfully led to expected results.
kidney cancer; kidney cancer genes; knockout mouse model; renal carcinoma therapeutics
陈金东,2000年于瑞典卡罗林斯卡医科大学分子医学系获得博士学位,现为美国罗切斯特大学医学中心肾癌研究室副教授和主任,美国癌症研究协会(AACR)资深会员。2003年克隆了NORE1和LSAMP两个肾癌相关基因,并在第94届AACR大会上获得Bristol-Myer Squibb奖。此后致力于肾癌敲基因小鼠模型的建立,现在已经建立了多个肾癌小鼠模型,其中一个FLCN肾近曲小管特异性敲基因小鼠模型是世界上唯一能产生各类肾癌细胞的肾近曲小管敲基因小鼠模型,并再次在第100届AACR大会上获得Sanofi-Aventis奖。至今已发表论文90余篇。
R737.1
A
1000-2715(2017)04-0347-11
[收稿2017-06-12;修回2017-07-12]
猜你喜欢
昆明医科大学学报(2021年10期)2021-12-02
科学大众(2021年6期)2021-07-20
健康必读·下旬刊(2020年11期)2020-11-11
学苑创造·A版(2020年9期)2020-10-13
保健与生活(2020年13期)2020-07-24
家庭医药(2019年8期)2019-08-27
保健与生活(2019年9期)2019-07-31
家庭科学·新健康(2019年4期)2019-05-05
娃娃乐园·综合智能(2019年3期)2019-04-03
中外医疗(2017年24期)2017-11-15
