
组织特异性蛋白质复合体的识别
2017-09-15 05:58:03丁霞张晓飞易鸣
数学杂志 2017年5期
丁霞,张晓飞,易鸣
(1.武汉大学数学与统计学院,湖北武汉430072)
(2.华中师范大学数学与统计学学院,湖北武汉430079)
(3.华中农业大学理学院,湖北武汉430070)
组织特异性蛋白质复合体的识别
丁霞1,张晓飞2,易鸣3
(1.武汉大学数学与统计学院,湖北武汉430072)
(2.华中师范大学数学与统计学学院,湖北武汉430079)
(3.华中农业大学理学院,湖北武汉430070)
本文研究了组织特异性蛋白质复合体的识别问题.利用蛋白质相互作用网络数据以及组织特异性基因表达数据构建组织特异性蛋白网络,利用多种代表性聚类算法对该网络进行聚类,并利用非负矩阵分解对聚类结果进行合并聚类,得到了组织特异性蛋白质复合体.结果表明,聚类效果得到明显提升,并且能识别出组织特异性蛋白质复合体.
蛋白质相互作用网络;复合体识别;组织特异性;非负矩阵分解
1 引言
在现如今的后基因组时代,对细胞间模块以及基因的关系进行系统分析和全面了解是一个非常重要的课题.随着生物信息学的高速发展,基因组学中大规模的高通量技术,如基于质谱的串联亲和纯化[1,2]、酵母双杂交[3,4]以及蛋白芯片技术为我们提供了海量的大规模生物网络,也为我们对生物网络进行系统的分析创造了可能.
众所周知,蛋白质很少单独行动,它们往往结合在一起形成复合体在生命体中进行生物功能[5].蛋白质复合体的综合研究有助于揭示蛋白质-蛋白质相互作用网络的结构、预测蛋白质的功能,更有助于阐明各种疾病的细胞机制[6].经过10多年的快速发展,已经涌现出了许多基于不同聚类机理的蛋白质相互作用网络功能模块检验方法.
尽管在此方面已经有不少研究,但是这些方法主要关注静态的蛋白质相互作用网络,而忽略了蛋白质功能作用的动态变化及组织特异机制.幸运的是,DNA微阵列技术的出现,使数以千计的基因的差异表达的各种实验条件被同时且定量监视,它提供了许多有关于时间以及组织特异的信息[7].目前也有少许算法研究动态网络,并探测动态复合体,但还没有算法涉及到组织特异的复合体侦测.
本文通过结合组织特异性基因表达数据以及人类蛋白质相互作用网络构建出一系列组织特异性蛋白网络,尝试探索组织特异功能模块的研究.本文的主要方法为对所构建的组织特异性蛋白网络利用多种方法对其进行聚类,并对结果进行组装,最后使用非负矩阵分解模型对组装的结果进行有效合并.实验结果表明,本文的方法与其他聚类方法相比,在检测蛋白质复合体上结果更好.
因为组织特异性蛋白复合体对于理解生物学功能以及确定生物标志物和功能靶标十分重要[8],因此探索组织特异功能模块很有必要.
2 方法
在本节中,本文首先介绍如何构建组织特异性蛋白网络,随后介绍如何检测组织特异性复合体.
2.1 构建组织特异性蛋白网络
组织特异性蛋白网络是结合蛋白质相互作用网络以及组织特异性基因表达数据两者来构建的.给定一个PPI网络,可以用图G=(V,E)来表示[9],其中V包含|V|=N个蛋白质,而E包含|E|条边.图G可以表示成一个邻接矩阵A,其中若有一条边连接蛋白质i与j,则Aij=1,否则Aij=0,在这种情况下,识别蛋白质复合体这一问题就转化为点的聚类问题.组织特异性基因表达数据是这N个蛋白质在T个组织中的基因水平,可以用一个N×T维矩阵F表示.
本文将利用矩阵A以及矩阵F来构建组织特异性蛋白网络.若蛋白质i与j有相关关系,即Aij=1,并且在组织t中,蛋白质i与蛋白质j均显著表达,即Fit>0并且Fjt>0,则蛋白质i与蛋白质j在组织t中存在相关关系.根据上述方法,对T个组织进行构建,则可得到T个组织特异性蛋白网络.
2.2 识别组织特异性蛋白质复合体
在本节中,本文先对组织特异性蛋白质相关关系网络中的每一个组织分别使用基本聚类方法,并使用非负矩阵分解模型来合并相似组织特异性蛋白质复合物,得到新的复合体,算法的基本流程如图1所示.
2.2.1 基本聚类方法
本文首先利用7种基本的聚类方法分别对这T个组织特异蛋白网络进行聚类,构建蛋白质复合体,所用的7种方法分别为MCL、MCODE、MINE、ClusterONE、DPClus、SPICi、CoAch.
MCL是通过模拟在PPI网络中流的自由行走来检测蛋白质复合体的经典算法,它定义了指派节点概率的Expansion操作和改变节点游走概率的In fl ation操作来模拟随机游走的扩展和收缩行为[10,11].
MCODE是一种基于蛋白质的连接值来检测蛋白质复合体的计算方法,它首先利用节点的局部邻域密度给PPI网络中每个节点进行加权,然后选择具有最高权值的节点作为初始聚类的种子节点,并由种子节点向外扩张形成最后的簇(蛋白质模块)[11,14].
MINE是一种类似于MCODE的凝聚聚类算法,但它使用了一个改进的顶点加权策略,并且可以衡量网络模块性,而这两者都有助于避免使用生长群内包含的临界点来定义模块的边界[13].
DPClus是一种通过簇边界的跟踪进行聚类的算法,它不仅利用模块密度而且利用新定义的粗特性CP完成复合体检验[11,14].
ClusterONE是一种能识别带重叠的蛋白质复合体的一种算法,它依赖于重叠领域扩张[15].
CoAch是一种利用核心依附关系进行复合体检测的算法,该算法分为两个阶段,第1阶段从邻接图中定义核心顶点,然后从中检测蛋白质复合体的核心蛋白质,第2阶段为将附属蛋白质逐个连接到核心蛋白质所代表的复合体中[11,16].
SPICi是一种高效算法,SPICi种子集群根据其加权度的节点,如果支撑足够高,并且集群的密度低于用户定义的阈值,则此非集群节点将会添加到集群中,否则,群集被输出,这个簇的节点将会从网络中移除[17].
2.2.2 非负矩阵分解模型
对每一个组织,分别使用上述7种聚类方法,可以得到7个复合体矩阵V1,V2,···,V7,Vi(i=1:7)为N×Pi(i=1:7)矩阵,其中N代表蛋白质的个数,Pi为第i种聚类方法所识别的蛋白质复合体的个数.对于矩阵Vi,若蛋白质Ni,Nj,···,Nk组成第e个复合体(1<=e<=Pi),则在第e列中,除了蛋白质Ni,Nj,···,Nk所对应的元素为1外,其余元素为0.将这7个复合体矩阵V1,V2,···,V7横向排列,得到矩阵V=[V1,V2,···,V7],V为N行P列的矩阵依造此方法,可构建出T个矩阵.
接着,我们使用了非负矩阵分解模型来合并相似瞬时蛋白质复合物.它提供了一种对非负矩阵的低秩逼近,并且已被广泛地运用到聚类当中[18,19].Lee和Seung的非负矩阵分解方法,设定模型为
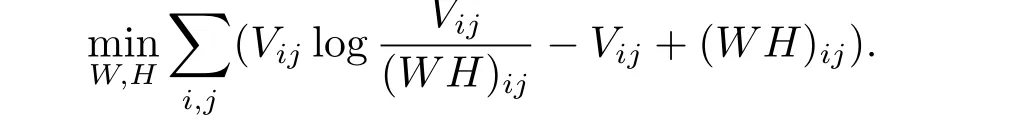
利用更新法则
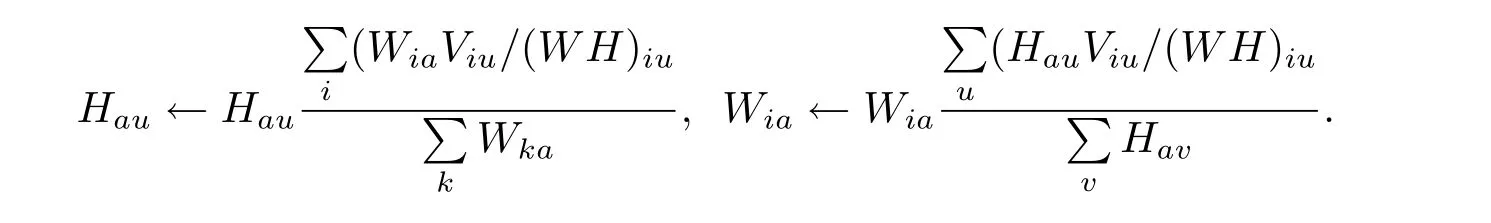
最后得到矩阵W(N×K)和H(K×P),本文只对矩阵W进行研究,将其横向归一,即令Uik=Wik/Wi..得到U之后,设定过滤阈值τ,若Uij>τ,则蛋白质Ni是复合体Kj的组成部分.由上可知,本次算法共有两个参数,所识别的复合体的个数K以及过滤阈值τ.由于复合体大多是由3个及3个以上的蛋白质组合而成,因此对所识别出的复合体进行过滤,将蛋白质个数<2的复合体舍去.
3 结果
3.1 构建组织特异性蛋白质子网络
本文从BIOGPS项目中的Af f ymetrix数据集中获得了83个人体组织和细胞系的转录水平[20],并从BioGrid网站[21]中下载到人体蛋白质-蛋白质相互作用关系,构建了83个组织特异性蛋白网络,具体处理数据以及构造方法详见文献[20],本文挑选了蛋白质对个数>10000的26个组织进行分析,这26个组织或者细胞分别为:BDCA 4+树突状细胞、支气管上皮细胞、CD105+内皮、CD19+B细胞、髓细胞、造血干细胞、CD4+T细胞、CD56+自然杀伤细胞、CD71+早期红细胞前体细胞、CD8+T细胞、心脏肌细胞、肠和直肠腺癌、慢性粒细胞性白血病k-562、早幼粒细胞性白血病淋巴细胞(MOLT-4)、白血病HL-60、淋巴瘤burkitt(Daudi)、淋巴瘤burkitt(Raji)、日间松果体、夜间松果体、前额叶皮层、视网膜、前列腺、平滑肌、甲状腺、全血.
3.2 黄金标准蛋白质复合体
为了衡量所检测出的复合体的精确性,本文选择了一个广泛使用的复合体标准作为黄金标准,该标准是从哺乳动物蛋白质复合体的CORUM[22]数据库中得到,最终获得由2151个蛋白质组成的324个复合体,本文中只选取其中蛋白质个数大于3个的复合体.
3.3 评价标准
我们将判断预测的复合体是否能很好地对应到已知的复合体作为评判标准.ACC[23]是用来测量几何精度的,在这项研究中,它被用来评估预测的复合体与参考的复合体之间的相似性.MMR(the Maximum Matching Ratio)由Paccanaro提出的用来评估相对于参考蛋白质复合体来说预测的蛋白质复合体是否符合期望的一个评价标准.

图2:参数τ在不同组织识别出不同复合体的曲线图
3.4 基本聚类方法的参数选择
MCL有一个用来调整聚类的间隔尺寸的参数,俗称膨胀率,本文设定其取值范围从3.0到5.0,步长为0.2;MCODE设定蛋白质个数为3,其余参数默认;MINE设定蛋白质个数为3,其余参数默认;DPCLUS有两个参数,最小密度d以及最小聚类性质参数cp,本文设定其值分别为0.7以及0.5;ClusterOne参数设为默认;CoAch有一个参数ω,用来过滤冗余的核心蛋白质,本文设定取值范围为0.225到0.925,步长为0.05;SPICi有两个参数,其中我们设定密度阈值这一参数的取值范围为0.1到1,步长为0.1.
对于以上7种算法,挑选出使得每种算法的ACC和MMR的调和平均数最大的参数值作为最后选定的参数值.
3.5 参数选择
本文的算法中,共有两个参数K以及τ,K为所识别的蛋白质复合体的个数,根据过往者的经验,设定其取值范围从600到1600,步长为200,τ为过滤阈值,设置其取值范围为0到0.9,步长为0.1.在对26个组织分别进行上述算法后,得到表1.

表1:ACC-结果比较
在对所有组织计算中发现,一般复合体个数在600-2000并且阈值在0或者0.1的情况下表现良好,由于篇幅有限,仅挑选出4个组织进行参数分析,分别为:甲状腺、B细胞、前额叶皮层、T细胞,如图2.
3.6 与其他聚类方法进行比较分析
在这一章中,我们将本文的算法与其他7种算法对这26个组织或者细胞的蛋白质网络进行聚类之后的结果进行比较.对于其他7种基本聚类方法,我们取其ACC和MMR的调和平均数为这26个组织最后的结果,从表1中可以看出,本文的算法最后得到的ACC值在24个组织中处于最大值,两个组织中居于第二.
本文将26个组织所用的7种方法得到的最高值与本文所用的方法进行比较,提升最高的组织是前列腺,提高值为13%.在与其他7种方法分别单独比较时,提高最高的百分比分别为:51.61%、33.33%、39.53%、122.22%、27.03%、25.00%、27.91%,具体提升情况可参见图3,从图中我们可以看出,MCODE算法所得到的结果最差,在26个组织中,使用非负矩阵分解得到的结果均比其提高30%以上;其次是MCL,提高了8%到40%;而CLusterONE表现最好,有两个组织比本文的算法分别高出1.96%、3.08%.

图3:与其他组织比较,NMF提升百分比的频数图
从上述结果中可以看出,本文所提出的算法与其他7种方法相比是具有优越性的.
4 讨论与分析
组织特异性蛋白质复合体对于理解生物学功能以及确定生物标志物和功能靶标十分重要,这也是本文的研究动机.同一个蛋白质在不同的组织中会与不同的蛋白质相结合,举例来说,转运蛋白1(TNPO1)在树突状细胞中与蛋白质CD4、PPP3CA、TNPO3结合,在髓细胞中与SRP19、TNPO3相结合,而在平滑肌中则与蛋白质IPO5、IPO7、NUTF2、RAN、SRP19结合形成复合体,由此可以看出在不同的组织中其会与不同的蛋白质相结合,而TNPO1与TNPO3则同时出现在不同组织的同一个复合体中,这与生命活动也是相符合的.
在真正的生命活动中,蛋白质会在不同的组织中与不同的蛋白质相结合,而许多现有的检测蛋白质复合物模型都是在静态PPI网络模型中直接检测,而忽略了蛋白质复合体的空间特异性.本文利用多种方法对不同的组织构建组织特异性蛋白质相互作用网络,并使用非负矩阵分解模型对其他聚类结果进行合并聚类,并在获取组织特异性蛋白质复合体时得到了良好的结果.同时,本文也有一些不足,虽然本文的结果在ACC标准中表现良好,但在MMR这一标准中仍需改进,同时,本文仅选取一个黄金标准复合体,在接下来的工作中,我们可以参考多组黄金标准复合体进行方法之间的比较.
[1]Aebersold R,Mann M.Mass spectrometry-based proteomics[J].Nature,2003,422(6928):198-207.
[2]Ho Y,Gruhler A,Heilbut A,et al.Systematic identif i cation of protein complexes in Saccharomyces cerevisiae by mass spectrometry[J].Nature,2002,415(6868):180-183.
[3]Ito T,Chiba T,Ozawa R,Yoshida M,Hattori M,Sakaki Y.A comprehensive two-hybrid analysis to explore the yeast protein interactome[J].Proceed.National Acad.Sci.United States America, 2001,98(8):4569-4574.
[4]Uetz P,Giot L,Cagney G,Mansf i eld T A,et al.A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae[J].Nature,2000,403(6770):623-627.
[5]Gavin A C,B sche M,Krause R,et al.Functional organization of the yeast proteome by systematic analysis of protein complexes[J].Nature,2002,415(6868):141-147.
[6]Lage K,Karlberg E O,Størling Z M,et al.A human phenome-interactome network of protein complexes implicated in genetic disorders[J].Nature Biotechnology,2007,25(3):309-316.
[7]Lo K,Raftery A E,Dombek K M,et al.Integrating external biological knowledge in the construction of regulatory networks from time-series expression data[J].BMC Sys.Bio.,2012,6(2):101.
[8]Vasmatzis G,Klee E W,Kube D M,Therneau T M,Kosari F.Quantitating tissue specif i city of human genes to facilitate biomarker discovery[J].Bioinformatics,2007,23(11):1348-1355.
[9]Li D,Li J,Ouyang S,Wang J,Wu S,Wan P,Zhu Y,Xu X,He F.Protein interaction networks of Saccharomyces cerevisiae,Caenorhabditis elegans and Drosophila melanogaster:large-scale organization and robustness[J].Proteomics,2006,6(2):456-461.
[10]Enright A J,Dongen S V,Ouzounis C A.An efficient algorithm for largescale detection of protein families[J].Nucleic Acids Res,2012,30(7):1575-1584.
[11]冀俊忠,刘志军,刘红欣,刘椿年.蛋白质相互作用网络功能模块检测的研究综述[J].自动化学报,2014, 40(4):577-593.
[12]Bader G D,Hogue C W V.An automated method for f i nding molecular complexes in large protein interaction networks[J].BMC Bioinformatics,2003,4(1):2.
[13]Rhrissorrakrai K,Gunsalus K C.MINE:module identif i cation in networks[J].BMC Bioinformatics, 2011,12(1):192.
[14]Altaf-Ul-Amin M,Shinbo Y,Mihara K,Kurokawa K,Kanaya S.Development and implementation of an algorithm for detection of protein complexes in large interaction networks[J].BMC Bioinformatics,2006,7(1):207.
[15]Nepusz T,Yu H,Paccanaro A.Detecting overlapping protein complexes in protein-protein interaction networks[J].Nature Methods,2012,9(5):471-472.
[16]Wu M,Li X L,Kwoh C K,Ng C K.A core-attachment based method to detect protein complexes in PPI networks[J].BMC Bioinformatics,2009,10(1):169.
[17]Jiang P,Singh M.SPICi:a fast clustering algorithm for large biological networks[J].Bioinformatics, 2010,26(8):1105-1111.
[18]Lee D D,Seung H S.Learning the parts of objects by non-negative matrix factorization[J].Nature, 1999,401(6755):788-791.
[19]Ding C,He X F,Simon H D.On the equivalence of nonnegative matrix factorization and spectral clustering[J].Siam Intern.Confer.Data Min.,2005,5:606-610.
[20]Lopes T J,Schaefer M,Shoemaker J,Matsuoka Y,Fontaine J F,Neumann G,Andrade-Navarro M A,Kawaoka Y,Kitano H.Tissue-specif i c subnetworks and characteristics of publicly available human protein interaction databases[J].Bioinformatics,2011,27(17):2414-2421.
[21]Chatr-aryamontri A,Breitkreutz B J,Heinicke S,et al.The Biogrid interaction database:2013 update[J].Nucleic Acids Research,2013,41(2):816-823.
[22]Havugimana P C,Hart G T,Nepusz T,et al.A census of human soluble protein complexes[J].Cell, 2012,150(5):1068-1081.
[23]Li X,Wu M,Kwoh C K,et al.Computational approaches for detecting protein complexes from protein interaction networks:a survey[J].BMC Genomics,2010,11(4):S3.
[24]Ou-Yang L,Dai D Q,Zhang X F.Protein complex detection via weighted ensemble clustering based on bayesian nonnegative matrix factorization[J].Plos One,2013,8(5):639-642.
[25]Ou-Yang L,Dai D Q,Li X L,Wu M,Zhang X F,Yang P.Detecting temporal protein complexes from dynamic protein-protein interaction networks[J].BMC Bioinformatics,2014,15(1):16001-16005.
[26]Zhang X F,Dai D Q,Ou-Yang L,Yan H.Detecting overlapping protein complexes based on a generative model with functional and topological properties[J].BMC Bioinformatics,2014,15(2):836-842.
[27]Zhang W,Zou X F.A new method for detecting protein complexes based on the three node cliques[J]. IEEE/ACM Trans Comput.Biol.Bioinform,2015,12(4):879-886.
[28]涂俐兰.两两序列比对的一种新方法[J].数学杂志,2006,26(1):67-70.
IDENTIFICATION OF THE TISSUE SPECIFIC PROTEIN COMPLEXES
DING Xia1,ZHANG Xiao-fei2,YI Ming3
(1.School of Mathematics and Statistics,Wuhan University,Wuhan 430072,China)
(2.School of Mathematics and Statistics,Central China Normal University,Wuhan 430079,China)
(3.School of Science,Huazhong Agricultural University,Wuhan 430070,China)
In this paper,we study the identif i cation problem of tissue-specif i c protein complexes.By using a variety of typical clustering algorithm to cluster the network,we construct a tissue-specif i c protein-protein interaction network based on the protein-protein interaction networks as well as the tissue-specif i c gene expression data,then merge the results with non-negative matrix factorization model to obtain tissue-specif i c protein complexes.The results show that clustering ef f ect has been signif i cantly improved,and can identify tissue-specif i c protein complexes.
protein-protein interaction networks;complexes identif i cation;tissue-specif i c; non-negative matrix factorization
O212.4;O212.5
A
0255-7797(2017)05-1093-08
2015-01-06接收日期:2015-05-06
国家自然科学基金资助(11275259);国家自然科学基金资助(91330113).
丁霞(1990-),女,湖北鄂州,硕士,主要研究方向:生物信息.
2010 MR Subject Classif i cation:92B05
猜你喜欢
电子测试(2017年15期)2017-12-18 07:19:27
中国医疗保险(2017年5期)2017-05-17 08:26:39
三峡大学学报(自然科学版)(2016年6期)2016-04-16 05:02:56
智能系统学报(2015年4期)2015-12-27 09:38:39
中国康复理论与实践(2015年10期)2015-12-24 05:42:46
原子与分子物理学报(2015年1期)2015-11-24 12:49:26
现代电生理学杂志(2015年1期)2015-07-18 11:02:16
电子设计工程(2015年6期)2015-02-27 12:04:53
华东师范大学学报(自然科学版)(2014年6期)2014-02-27 13:40:55
食品科学(2013年15期)2013-03-11 18:25:51

- 数学杂志的其它文章
- BIFURCATION IN A RATIO-DEPENDENT PREDATOR-PREY SYSTEM WITH STAGE-STRUCTURED IN THE PREY POPULATION
- FORCING AN ω1-REAL WITHOUT ADDING A REAL
- 面板数据分位数回归模型的参数估计与变量选择
- ON A NEW NONTRIVIAL ELEMENT INVOLVING THE THIRD PERIODICITY γ-FAMILY IN π∗S
- CONVERGENCE THEORY ON QUASI-PROBABILITY MEASURE
- 两类二元函数芽的一个共同性质及其应用