
酰胺酶催化机制研究进展
2017-09-12 10:45金建强吴哲明郑仁朝
发酵科技通讯 2017年3期
金建强,吴哲明,郑仁朝
(浙江工业大学 生物工程学院,浙江 杭州 310014)
酰胺酶催化机制研究进展
金建强,吴哲明,郑仁朝
(浙江工业大学 生物工程学院,浙江 杭州 310014)
酰胺酶是一类主要作用于分子内C—N键,催化酰胺水解生成相应的羧酸和氨的水解酶,在自然界中广泛存在.根据酰胺酶氨基酸序列,可将其分为腈水解酶家族和酰胺酶标签家族两类.综述了近年来对上述两类酰胺酶结构和催化机制方面的研究进展,并着重阐述了数个不同来源酰胺酶的具体催化过程.腈水解酶家族成员的催化三联体为Cys-Lys-Glu,其催化过程比较一致,Cys主要负责亲核进攻,Glu作为广义碱参与催化反应,Lys则负责稳定过渡态.酰胺酶标签家族酰胺酶的催化三联体为Ser-cisSer-Lys,但不同来源的酰胺酶之间的催化机理差异较大,依据Lys所扮演的角色可以将其催化机制分为广义酸催化和广义碱催化两种模式.
酰胺酶;腈水解酶家族;酰胺酶标签家族;催化机制
酰胺酶(Amidase,EC 3.5.1.x)又称酰胺水解酶(Amidohydrolase),是一类能够催化酰胺键断裂生成相应羧酸和氨的水解酶,广泛存在于细菌(广谱酰胺酶)、霉菌(青霉素酰化酶)、真菌(甲酰胺酶,乙酰胺酶以及广谱酰胺酶)、酵母(烟酰胺酶以及广谱酰胺酶)、植物(肽酶)和动物(花生四烯酸乙醇胺酰胺酶)等[1-2]生物体中.Claridge等[3]曾于1960年报道了青霉素酰化酶(Penicillin amidase).随后,Kelly和Jakoby于1964年分别从Pseudomonasaeruginosa和Pseudomonasfluorescens中发现了酰胺酶[4-5].绝大部分酰胺酶的底物谱广,能高效催化各种脂肪族、芳香族以及杂环族酰胺的水解.酰胺酶因具有良好的化学和立体选择性,在手性羧酸、酰胺衍生物和光学纯氨基酸等手性化合物的合成中极具潜力,正日益受到工业界的重视.
随着对酰胺酶研究的不断深入,其分子结构和催化机制成为了研究者关注的重点.分析多种酰胺酶之间的结构及其催化机理有助于系统并深入地理解其空间结构与功能的关系.近年来,关于酰胺酶结构和催化机制的研究已经有了一定的进展.笔者主要阐述了一些酰胺酶的结构和催化机制,为酰胺酶的催化反应研究提供理论基础.
1 酰胺酶的分类及结构
根据酰胺酶的氨基酸序列,可以将其大致分为两类[2,6]:腈水解酶家族(Nitrilase superfamily)酰胺酶,其催化三联体为Cys-Lys-Glu;酰胺酶标签(Amidase signature,AS)家族酰胺酶,以Lys-Ser-Ser为催化三联体,在其一级结构中含有一段约130 个保守氨基酸残基且富含GGSS序列片段的特征序列(标签序列)[7].
腈水解酶家族酰胺酶是一类巯基酶,含有保守的亲核半胱氨酸[8].腈水解酶家族酰胺酶的底物谱较窄,通常只能催化脂肪族酰胺水解.该家族酰胺酶和腈水解酶具有序列相似性,其与腈水解酶可能存在进化上的相关性.腈水解酶家族酰胺酶多重序列比对结果表明,其一级结构含有许多保守片段,并且与腈水解酶、腈水合酶和β-丙氨酸合成酶同源.腈水解酶家族成员的四级结构通常为同源四聚体或同源六聚体.其单体一般呈α-β-β-α式夹心折叠结构,并且有一个高度保守的催化三联体Cys-Lys-Glu负责共价键催化(图1),其中Cys为亲核体[9].有关腈水解酶家族酰胺酶的晶体结构很少,已报道的该家族酰胺酶主要来源于Geobacilluspallidus[9-11],P.aeruginosa[12],Agrobacteriumsp.[13]和Helicobacterpylori[14]等.图1中:Agrobacterium为N-carbamyl-D-amino acid amidohydrolase (DCase) A链,来源于Agrobacteriumsp. KNK712,PDB ID code: 1ERZ;Helicobacter为Formamidase (AmiF) A链,来源于Helicobacterpylori,PDB ID code: 2DYU;Geobacillus为Geobacilluspallidus酰胺酶,来源于GeobacilluspallidusRAPc8,PDB ID code: 2PLQ;Pseudomonas为Aliphatic amidase,来源于Pseudomonasaeruginosa,GenBank accession No. KSP66314.1;催化三联体Cys-Lys-Glu以▼标记.
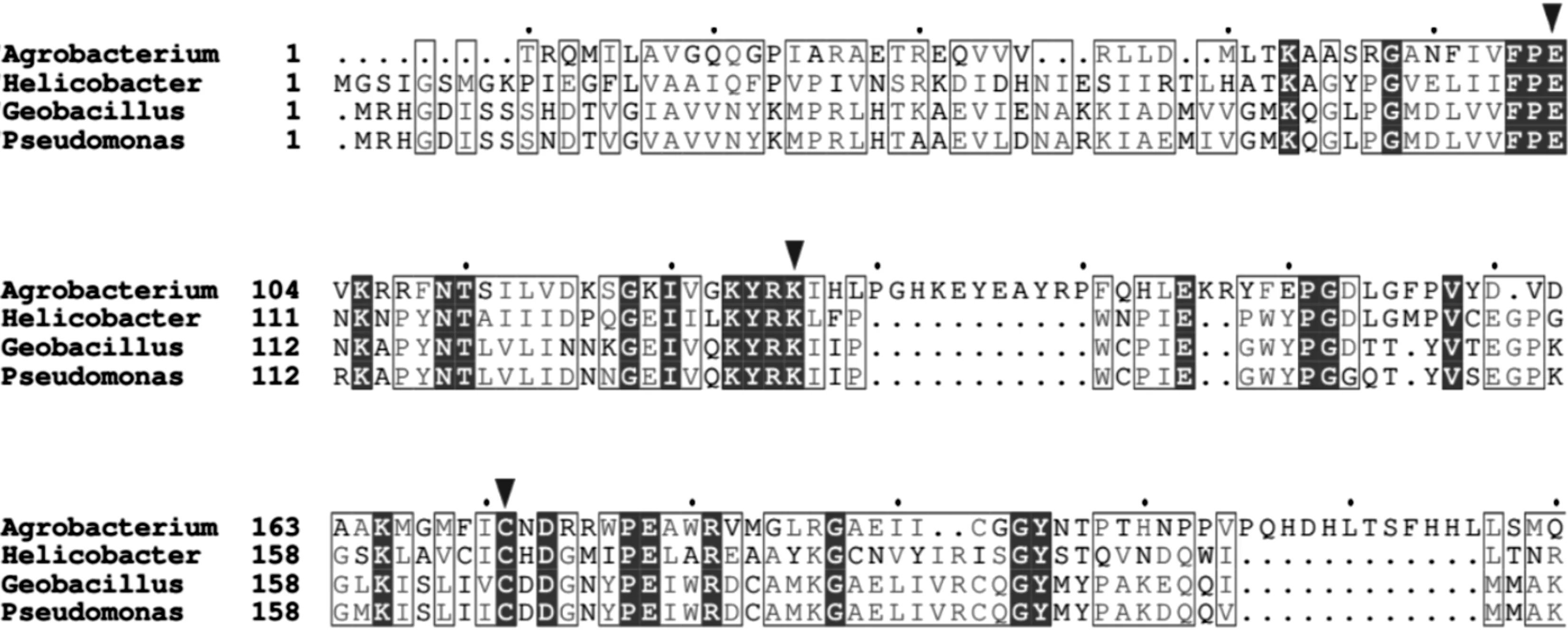
图1 腈水解酶家族酰胺酶序列比对Fig.1 Amino acid sequence alignment of nitrilase superfamily
酰胺酶标签家族酰胺酶与腈水解酶家族成员有明显的区别.该家族酰胺酶通常有较宽的底物谱,可催化如脂肪族酰胺、芳香族酰胺、杂环类酰胺和α-取代酰胺等的水解.其四级结构与腈水解酶家族成员不同,一般呈同源二聚体或同源八聚体[8,15-16].标签家族成员的催化三联体并非典型的丝氨酸水解酶类结构(Ser-Asp-His)[7],而是Ser-cisSer-Lys.其中Ser作为亲核体通过其侧链上的羟基氧(Oγ)对底物的羰基发动亲核进攻,而cisSer为不常见的顺式结构,处于GGSS指纹序列的中心位置(图2),并与Lys一起组成了质子传递网络.此外,亲核体Ser以及与其相连的2~3 个保守氨基酸(通常为Gly,Thr或Asp)构成了活性中心的发卡环结构,参与氧负离子洞的形成.目前关于酰胺酶标签家族酰胺酶的结构报道包括Malonamidase E2 (MAE2)[7,17],Glu-tRNAGln-dependent amidotransferase (GatCAB)[18],6-aminohexanoate cyclic dimer hydrolase (NylA)[19],Fatty acid amide hydrolase (FAAH)[20-22],Peptide amidase (Pam)[23]和Aryl acylamidase (AAA)[24]等.图2中:PAM为Peptide amidase,来源于Stenotrophomonasmaltophilia,GenBank acession No. CAC93616;MAE2为Malonamidase E2,来源于Bradyrhizobiumjaponicum,GenBank acession No. AAD01507;NylA为6-aminohexanoate cyclic dimer hydrolase,来源于Arthrobactersp. KI72,PDB ID code: 3A2P;AAA为Aryl acylamidase,来源于某土壤细菌CSBL00001,PDB ID code: 4YJI;FAAH为Fatty acid amide hydrolase,来源于Rattusnorvegicus,PDB ID code: 2VYA;催化三联体Ser-cisSer-Lys以▼标记.

图2 酰胺酶标签家族酰胺酶序列比对Fig.2 Amino acid sequence alignment of amidase signature family
此外,绝大多数酰胺酶不含金属离子,但有一些研究者报道了Rhodococcussp.[25],KlebsiellapneumoniaeNCTR 1[26]和BrevundimonasdiminutaTPU 5720[27]的酰胺酶的活性位点上含有Co2+和/或Fe3+.
2 酰胺酶的催化机制
酰胺酶可以催化包括酰胺水解、酰基转移、酸转移、酯水解和酯转移等在内的多种反应,但以酰胺水解和酰基转移反应的活性最高[1-2].当反应体系存在羟胺时,由于羟胺具有比水更强的亲核性,底物酰胺会优先和羟胺发生酰基转移反应.1986年,Maestracci等[28]基于此特性研究了Brevibacteriumsp. R312酰胺酶的酰基转移反应.他们以乙酰胺和羟胺为双底物,发现该反应符合“Bi-Bi Ping-Pong”机制.在酰基转移反应中,酰胺酶首先与底物酰胺结合形成酶-底物-酰基中间体,同时释放出氨;随后亲核试剂羟胺与中间体反应生成氧肟酸;最后氧肟酸脱离,酰胺酶恢复为初始状态.随后,Kobayashi等[29]提出了酰胺酶的催化机制,他们认为底物酰胺的羰基在受到亲核进攻后,会与酶形成一个四面体中间体.中间体存在时间极短,其因氨的形成并脱离而快速转变为酰基-酶复合物.在水分子加入后,酰基-酶复合物发生水解,酶脱离并形成相应的酸.Fournand等[2]通过对酰胺酶的酰胺水解反应以及酰基转移反应研究,同样确认了两者反应过程均符合“Bi-Bi Ping-Pong”机制.酰胺酶酰基转移反应和水解反应的“Bi-Bi Ping-Pong”机制[1-2]为

2.1 腈水解酶家族酰胺酶的催化机制
有关腈水解酶家族酰胺酶催化机理的研究较少,已报道的主要集中于N-carbamyl-D-amino acid amidohydrolase (DCase),Formamidase (AmiF)和G.pallidusRAPc8 aliphatic amidase等少数几个酶中.从这几个酶来看,腈水解酶家族酰胺酶的催化过程比较一致:催化三联体中,Cys负责亲核进攻,处于去质子化状态的Glu作为广义碱,增强Cys的亲核性并负责质子的传递,而Lys则是用于稳定催化过程中的过渡态,并不直接参与质子传递.Nakai等[13]对来源于Agrobacteriumsp. KNK712的N-氨甲酰基-D-氨基酸酰胺水解酶(DCase)进行了晶体结构解析,确认其催化三联体为Cys171-Lys126-Glu46,并提出了该酶的具体催化过程:
1) Glu46处于去质子化状态,直接与Cys171相互作用,夺取后者巯基上的质子,激活其亲核进攻能力.活化的Cys171进攻底物的羰基碳,形成带氧负离子的酶-底物复合体,并被Lys126所稳定.
2) 复合体的氨基氮夺取Glu46的质子变为氨分子而脱离,羰基重新形成,构成酰基-酶中间体,Glu46恢复去质子化状态.
3) 水分子取代氨分子进入反应中心,被Glu46夺去一个质子后,成为活化状态,进攻酰基-酶中间体并与之共价结合,而羰基再次解体产生氧负离子.
4) 中间体解体,Cys171脱离并获得Glu46的质子,恢复稳定.羰基恢复,形成催化产物N-羧基氨基酸,并进一步自发水解为氨基酸和二氧化碳.
DCase的催化机制[13]为

另一种报道的腈水解酶家族成员为来源于H.pylori的甲酰胺酶(AmiF),AmiF的催化机制[14]为Hung等[14]通过对AmiF晶体结构的解析,认为该酶的具体催化步骤为:

1) 底物甲酰胺分子进入催化活性中心,去质子化的Glu60夺取Cys166的巯基质子,激活其亲核进攻反应.Cys166与底物共价结合,形成带氧负离子的酶-底物复合体,并被Lys133和His167通过氢键所稳定.
2) 底物氨基获得Glu60的质子形成氨被释放,复合体分解,羰基恢复,构成酰基-酶中间体.
3) 水分子进入反应中心,经Glu60活化后进攻酰基-酶中间体.中间体分解,羰基恢复,形成产物甲酸,Cys166从Glu60处获得质子,三联体恢复初始状态.
2.2 酰胺酶标签家族酰胺酶的催化机理

2.2.1 去质子化Lys介导的广义碱催化模式
脂肪酸酰胺水解酶(FAAH)是一种来源于哺乳动物的内源性大麻素降解酶,属于典型的酰胺酶标签家族酰胺酶,关于其结构研究的报道较多[31-36].Patricelli等[36-37]研究结果表明:催化初始阶段,FAAH的Lys142并未处于质子化状态,而是作为广义碱接受来自cisSer217的质子.其具体催化步骤[31-33]如下:
1) 处于去质子化状态的Lys142首先夺取cisSer217的质子,失去质子的cisSer217则夺取Ser241的质子,激活Ser241的亲核能力.
2) 活化的Ser241进攻底物中的羰基碳并与之共价结合,羰基解体,产生氧负离子(由Gly240-Gly239-Ile238构成的氧负离子洞稳定),形成酶-底物复合体.与此同时,cisSer217将其质子贡献给氨基氮,并获得Lys142的质子补偿,两者恢复去质子化状态.
3) 酰胺底物的C—N键断裂,形成胺脱离复合体,羰基重新恢复,形成酰基-酶中间体.随后,Lys142再次质子化,获得cisSer217的羟基氢.而失去质子的cisSer217作为广义碱夺取水分子的一个质子,并激活水分子的亲核进攻能力.活化的水分子(即HO-部分)进攻酰基-酶中间体的羰基碳,羰基再次解体并产生氧负离子,形成新的四面体中间体.
4) 最后,中间体分解、羰基恢复,形成第二个产物——羧酸.Ser241脱离后获得cisSer217的质子,而后者则得到Lys142的质子补偿,三联体恢复初始状态.
酰胺酶FAAH催化机制[33]为

芳基酰基酰胺酶(AAA)以Ser187,Ser163与Lys84作为催化三联体,其中Lys84同样是作为广义碱角色,接受来自Ser163的质子,并辅助Ser163活化亲核体Ser187[24].但其催化步骤与FAAH并不完全相同,主要包括:
1) 在碱性环境(pH>10)下的Lys84处于去质子化状态,其通过质子传递网络获得cisSer163的质子,并使之转而夺取亲核体Ser187上的质子,激活Ser187对底物的亲核进攻反应.
2) 去质子化的Ser187与底物(对乙酰氨基酚)的羰基碳共价结合,氧负离子则由发卡环上的两个氨基酸残基(Gly185和Gly184)通过氢键稳定,构成四面体中间体.
3)cisSer163质子化底物的亚氨基,形成对氨基苯酚脱离催化体系.失去质子的cisSer163直接夺取水分子上的质子,并激活其对中间体的脱酰反应.
4) 活化的水分子进攻中间体并与之结合,导致Ser187脱离,中间体解体,羰基恢复,形成羧酸.Ser187则夺取cisSer163的质子,后者则拿回在Lys84上的质子.最终酶与产物分离,酰胺酶获得再生.
酰胺酶AAA的催化机制[24]为

2.2.2 质子化Lys介导的广义酸催化模式
Labahn等[23]测定了Stenotrophomonasmaltophilia的肽酰胺酶Pam的四级结构,确定其催化三联体为Ser226-cisSer202-Lys123.他们认为Pam的Lys123在催化起始阶段就处于质子化状态,作为广义酸参与反应,并据此提出了相应的催化机制.该假设得到了动力学模拟研究结果的支持[30].Pam的具体水解反应过程为:
1) 处于质子化状态的Lys123降低了cisSer202的亲核性,但同时增强了cisSer202对底物酰胺羰基氧的质子化能力.Ser226作为亲核体首先进攻底物酰胺的羰基碳原子,同时cisSer202质子化羰基氧.随后,失去质子的cisSer202夺取Ser226的质子,形成酶-底物复合体.
2) 复合体上的氨基夺取cisSer202的质子,失去质子的cisSer202转而攻击带正电的Lys123并获得质子补偿,恢复稳定.
3) 质子化的氨基形成氨脱离复合体.Lys123重新夺回cisSer202上的质子,而cisSer202则获得底物上的羟基质子,两者恢复原来状态.同时,底物的羰基也重新产生,形成酶-酰基中间体.
4) 随后,与Ser226通过氢键连接的水分子进攻酶-酰基中间体使其分解,形成产物羧酸.同时Ser226获得水分子上的一个质子,重新恢复稳定状态.
酰胺酶PAM的催化机制[23]为

Shin等[7,17]对丙二酰胺酶MAE2的研究表明,其Lys62同样处于质子化状态.但与Pam有所区别,MAE2的亲核体Ser155由于受到Arg158的胍基的影响,其pKa降低,因此他们认为亲核体Ser155在催化初始阶段就处于去质子化状态(Ser155—O-),并且通过与cisSer131形成两个氢键来保持稳定性.该酶的具体催化机制如下:
1) Lys62处于质子化状态,而Ser155处于去质子化状态.在该状态下,Ser131的亲核性被减弱,但其质子化底物酰胺羰基氧的能力得到加强.催化开始时,处于激活状态下Ser177侧链氧原子(Oγ-)攻击底物酰胺上的羰基碳,与之共价结合,形成携带氧负离子的酶-底物复合体,并被Thr152,Gly153,Gly154以及Ser155组成的氧负离子洞所稳定.
2) 紧接着,cisSer131作为酸催化剂将其侧链上的质子传递给复合体上的底物氨基使其质子化,形成氨脱离反应体系.同时,羰基恢复,最终形成酶-酰基中间体.失去质子的cisSer131则从质子化状态下的Lys80那里获得质子补偿.
3) 随后,水分子进入反应中心.Lys80重新拿回cisSer131上的质子,后者被活化,进攻水分子并获得质子补偿,恢复稳定状态.而失去质子的水分子则进攻酶-酰基中间体,使其水解并形成羧酸.而Ser155则脱离并恢复为去质子化状态.至此,MAE2完成一个底物的水解反应过程.
MAE2的催化机制[17]为

来源于Arthrobactersp. KI72的NylA的催化过程与MAE2基本类似,但初始状态的Ser174并未处于去质子化状态,需要Ser150的激活才能进行亲核进攻[19].
3 结 论
作为一类应用广泛的生物催化剂,酰胺酶近年来受到越来越多的关注,不断有各种酰胺酶的空间结构被鉴定和解析,其相应的催化反应机制也逐渐被揭示.空间结构与催化机制的阐明对于酰胺酶的开发、改造和应用具有重要的意义.根据已知的酰胺酶结构,通过分子对接和动力学等方法研究药物与酶的结合模式和作用关键残基,可以使药物改造具有更好的方向性和针对性.通过探究酰胺酶的结构和作用机理,有助于准确高效地利用基因重组技术和蛋白质工程等手段改造酰胺酶,提升其催化活力和稳定性,为酰胺酶的工业化应用奠定基础.然而,酰胺酶来源广泛、类型众多,其催化过程并不相同,目前还没有统一的催化机制来解释其反应过程.另外,一些金属依赖型酰胺酶的催化机制并未明确.因此,对酰胺酶催化机理的研究仍有待深入.
[1] SHARMA M, SHARMA N N, BHALLA T C. Amidases: versatile enzymes in nature[J]. Reviews in environmental science and bio/technology, 2009, 8(4): 343-366.
[2] FOURNAND D, ARNAUD A. Aliphatic and enantioselective amidases: from hydrolysis to acyl transfer activity[J]. Journal of applied microbiology, 2001, 91(3): 381-393.
[3] CLARIDGE C A, GOUREVITCH A, LEIN J. Bacterial penicillin amidase[J]. Nature, 1960, 187: 237-238.
[4] KELLY M, KORNBERG H L. Purification and properties of acyltransferases fromPseudomonasaeruginosa[J]. Biochemical journal, 1964, 93(3): 557-566.
[5] JAKOBY W B, FREDERICKS J. Reactions catalyzed by amidases: acetamidase[J]. Journal of biological chemistry, 1964, 239: 1978-1982.
[6] MAKHONGELA H S, GLOWACKA A E, AGARKAR V B, et al. A novel thermostable nitrilase superfamily amidase fromGeobacilluspallidusshowing acyl transfer activity[J]. Applied microbiology and biotechnology, 2007, 75(4): 801-811.
[7] SHIN S, LEE T H, HA N C, et al. Structure of malonamidase E2 reveals a novel Ser-cisSer-Lys catalytic triad in a new serine hydrolase fold that is prevalent in nature[J]. EMBO journal, 2002, 21(11): 2509-2516.
[8] NOVO C, TATA R, CLEMENTE A, et al.Pseudomonasaeruginosaaliphatic amidase is related to the nitrilase/cyanide hydratase enzyme family and Cys166 is predicted to be the active site nucleophile of the catalytic mechanism[J]. FEBS letters, 1995, 367(3): 275-279.
[9] KIMANI S W, AGARKAR V B, COWAN D A, et al. Structure of an aliphatic amidase fromGeobacilluspallidusRAPc8[J]. Acta crystallographica section D: biological crystallography, 2007, 63(10): 1048-1058.
[10] WEBER B W, KIMANI S W, VARSANI A, et al. The mechanism of the amidases: mutating the glutamate adjacent to the catalytic triad inactivates the enzyme due to substrate mispositioning[J]. Journal of biological chemistry, 2013, 288(40): 28514-28523.
[11] AGARKAR V B, KIMANI S W, COWAN D A, et al. The quaternary structure of the amidase fromGeobacilluspallidusRAPc8 is revealed by its crystal packing[J]. Acta crystallographica, 2006, 62(12): 1174-1178.
[12] FARNAUD S, TATA R, SOHI M K, et al. Evidence that cysteine-166 is the active-site nucleophile ofPseudomonasaeruginosaamidase: crystallization and preliminary X-ray diffraction analysis of the enzyme[J]. Biochemical journal, 1999, 340(3): 711-714.
[13] NAKAI T, HASEGAWA T, YAMASHITA E, et al. Crystal structure of N-carbamyl-D-amino acid amidohydrolase with a novel catalytic framework common to amidohydrolases[J]. Structure, 2000, 8(7): 729-737.
[14] HUNG Chiulien, LIU Jiahsin, CHIU Weichun, et al. Crystal structure ofHelicobacterpyloriformamidase AmiF reveals a cysteine-glutamate-lysine catalytic triad[J]. Journal of biological chemistry, 2007, 282(16): 12220-12229.
[15] CHEBROU H, BIGEY F, ARNAUD A, et al. Study of the amidase signature group[J]. Biochimica et biophysica acta, 1996, 1298(2): 285-293.
[16] MAYAUX J F, CERBELAUD E, SOUBRIER F, et al. Purification, cloning, and primary structure of a new enantiomer-selective amidase from aRhodococcusstrain: structural evidence for a conserved genetic coupling with nitrile hydratase[J]. Journal of bacteriology, 1991, 173(21): 6694-6704.
[17] SHIN S, YUN Y S, KOO H M, et al. Characterization of a novel Ser-cisSer-Lys catalytic triad in comparison with the classical Ser-His-Asp triad[J]. Journal of biological chemistry, 2003, 278(27): 24937-24943.
[18] NAKAMURA A, YAO M, CHIMNARONK S, et al. Ammonia channel couples glutaminase with transamidase reactions in GatCAB[J]. Science, 2006, 312(5782): 1954-1958.
[19] YASUHIRA K, SHIBATA N, MONGAMI G, et al. X-ray crystallographic analysis of the 6-aminohexanoate cyclic dimer hydrolase: catalytic mechanism and evolution of an enzyme responsible for nylon-6 byproduct degradation[J]. Journal of biological chemistry, 2010, 285(2): 1239-1248.
[20] MILENI M, JOHNSON D S, WANG Z, et al. Structure-guided inhibitor design for human FAAH by interspecies active site conversion[J]. Proceedings of the national academy of sciences of the United States of America, 2008, 105(35): 12820-12824.
[21] MIN X, THIBAULT S T, PORTER A C, et al. Discovery and molecular basis of potent noncovalent inhibitors of fatty acid amide hydrolase (FAAH)[J]. Proceedings of the national academy of sciences of the United States of America, 2011, 108(18): 7379-7384.
[22] PALERMO G, ROTHLISBERGER U, CAVALLI A, et al. Computational insights into function and inhibition of fatty acid amide hydrolase[J]. European journal of medicinal chemistry, 2015, 91: 15-26.
[23] LABAHN J, NEUMANN S, BÜLDT G, et al. An alternative mechanism for amidase signature enzymes[J]. Journal of molecular biology, 2002, 322(5): 1053-1064.
[24] LEE S, PARK E H, KO H J, et al. Crystal structure analysis of a bacterial aryl acylamidase belonging to the amidase signature enzyme family[J]. Biochemical and biophysical research communications, 2015, 467(2): 268-274.
[25] NAWAZ M S, KHAN A A, SENG J E, et al. Purification and characterization of an amidase from an acrylamide-degradingRhodococcussp.[J]. Applied and environmental microbiology, 1994, 60(9): 3343-3348.
[26] NAWAZ M S, KHAN A A, BHATTACHARAYYA D, et al. Physical, biochemical, and immunological characterization of a thermostable amidase fromKlebsiellapneumoniaeNCTR 1[J]. Journal of bacteriology, 1996, 178(8): 2397-2401.
[27] KOMEDA H, HARIYAMA N, ASANO Y. L-Stereoselective amino acid amidase with broad substrate specificity fromBrevundimonasdiminuta: characterization of a new member of the leucine aminopeptidase family[J]. Applied microbiology and biotechnology, 2006, 70(4): 412-421.
[28] MAESTRACCI M, THIERY A, ARNAUD A, et al. A study of the mechanism of the reactions catalyzed by the amidaseBrevibacteriumsp. R312[J]. Agricultural and biological chemistry, 1986, 50(9): 2237-2241.
[29] KOBAYASHI M, FUJIWARA Y, GODA M, et al. Identification of active sites in amidase: evolutionary relationship between amide bond-and peptide bond-cleaving enzymes[J]. Proceedings of the national academy of sciences of the United States of America, 1997, 94(22): 11986-11991.
[31] LABAR G, MICHAUX C. Fatty acid amide hydrolase: from characterization to therapeutics[J]. Chemistry & biodiversity, 2007, 4(8): 1882-1902.
[32] MCKINNEY M K, CRAVATT B F. Structure and function of fatty acid amide hydrolase[J]. Annual review of biochemistry, 2005, 74: 411-432.
[33] MILENI M, KAMTEKAR S, WOOD D C, et al. Crystal structure of fatty acid amide hydrolase bound to the carbamate inhibitor URB597: discovery of a deacylating water molecule and insight into enzyme inactivation[J]. Journal of molecular biology, 2010, 400(4): 743-754.
[34] MCKINNEY M K, CRAVATT B F. Evidence for distinct roles in catalysis for residues of the serine-serine-lysine catalytic triad of fatty acid amide hydrolase[J]. Journal of biological chemistry, 2003, 278(39): 37393-37399.
[35] PATRICELLI M P, CRAVATT B F. Clarifying the catalytic roles of conserved residues in the amidase signature family[J]. Journal of biological chemistry, 2000, 275(25): 19177-19184.
[36] PATRICELLI M P, LOVATO M A, CRAVATT B F. Chemical and mutagenic investigations of fatty acid amide hydrolase: evidence for a family of serine hydrolases with distinct catalytic properties[J]. Biochemistry, 1999, 38(31): 9804-9812.
[37] PATRICELLI M P, CRAVATT B F. Fatty acid amide hydrolase competitively degrades bioactive amides and esters through a nonconventional catalytic mechanism[J]. Biochemistry, 1999, 38(43): 14125-14130.
(责任编辑:朱小惠)
Progress on the catalytic mechanism of amidase
JIN Jianqiang, WU Zheming, ZHENG Renchao
(College of Biotechnology and Bioengineering, Zhejiang University of Technology, Hangzhou 310014, China)
Amidases are a class of hydrolases, which catalyze the hydrolysis of C—N bonds of various amides to the corresponding carboxylic acids and ammonia. They are ubiquitous in many different types of organisms. According to their amino acid sequences, amidases can be assigned into nitrilase superfamily and amidase signature family. Here, we systematically review the progress of study in molecular structures and catalytic mechanisms of amidases, as well as the specific catalytic processes of specific amidases. All members of the nitrilase superfamily possess the conserved catalytic triad Cys-Lys-Glu, and exhibit similar catalytic processes. The residue Cys is responsible for the nucleophilic attack, Glu acts as a general base, while Lys is involved in the stabilization of the transition states. As for amidase signature family, the conserved catalytic triad is Ser-cisSer-Lys, and the catalytic processes vary from different signature amidases. According to the role of Lys, the catalytic mechanism can be divided into two types: general acid catalysis and general base catalysis.
amidase; nitrilase superfamily; amidase signature family; catalytic mechanism
2017-04-29
国家自然科学基金资助项目(21602199)
金建强(1991—),男,浙江台州人,硕士研究生,研究方向为生物催化与酶工程,E-mail: jinjq6967@163.com. 通信作者:郑仁朝教授,E-mail: zhengrc@zjut.edu.cn.
Q556.4
A
1674-2214(2017)03-0170-08
猜你喜欢
生物信息学(2022年1期)2022-04-01
分析科学学报(2018年3期)2018-09-03
科技创新导报(2018年1期)2018-05-07
石油化工应用(2018年3期)2018-03-24
中国洗涤用品工业(2017年2期)2017-04-16
西安文理学院学报(自然科学版)(2016年4期)2016-12-19
China International Studies(2016年3期)2016-07-14
当代化工研究(2016年2期)2016-03-20
中国洗涤用品工业(2016年2期)2016-02-28
质谱学报(2015年5期)2015-03-01
