
利用2b-RAD测序结合HRM分析技术开发与梨矮生性状相关的DNA分子标记
2017-09-11 09:04肖玉雄王彩虹田义轲杨绍兰李鼎立张海月
中国农业科学 2017年15期
肖玉雄,王彩虹,田义轲,杨绍兰,李鼎立,张海月
利用2b-RAD测序结合HRM分析技术开发与梨矮生性状相关的DNA分子标记
肖玉雄,王彩虹,田义轲,杨绍兰,李鼎立,张海月
(青岛农业大学园艺学院/青岛市园艺植物遗传改良与育种重点实验室,山东青岛 266109)
【目的】果树的矮生性状是一种重要的农艺性状,对果树的集约化栽培具有重要意义。来自西洋梨实生变异品种‘Le Nain Vert’的矮生性状受控于一个单显性基因,目前关于该基因的序列信息等还不清楚。本研究的目的是开发与其紧密连锁的DNA分子标记,为鉴定该基因提供依据。【方法】根据分离群体分组分析的原理,以‘矮生梨’ב茌梨’和‘2-3’ב绿宝石’2个F1杂交分离群体为试材,应用IIB型限制性内切酶的RAD技术(Restriction association site DNA,2b-RAD)对2对矮生型/普通型对比基因池进行基因组测序分析,在对比基因池间筛选出单核苷酸多态性(Single nucleotide polymorphism,SNP)位点,从中筛查出位于定位染色体上的SNPs,用高分辨率熔解曲线分析技术(High-resolution melting analysis,HRM)在群体上进一步检测验证,以确定其与位点的连锁关系。【结果】对4个样本(即4个对比基因池)的2b-RAD标签测序文库的测序结果共产生67 186 260条reads,平均每个样本测序reads数为16 796 565。将原始reads进行质量过滤后的统计结果表明,每个样本获得平均unique标签数目为86 810,平均测序深度为77×,该测序深度能够达到准确分型的标准。SOAP软件定位结果表明,4个测序文库中含有酶切位点的高质量reads占测序原始reads的70%以上,表明测序质量较好。在来自2个不同群体的矮生型基因池与普通型基因池间进行对比分析,初步筛选出SNP位点1 317个,其中有8个位于的定位染色体scaffold00074上。用HRM技术对这8个SNP标记在群体上的进一步检测结果表明,在‘矮生梨’ב茌梨’群体上有2个SNP标记、在‘2-3’ב绿宝石’群体上有4个SNP标记表现出与位点共分离的特性,根据其扩增子的熔解曲线形状差异,可有效区分矮生型和普通型表型。在来自‘矮生梨’ב茌梨’群体的215个杂种后代和来自‘2-3’ב绿宝石’群体的168个杂种后代中,未发现有标记与性状的重组类型。【结论】2b-RAD测序技术与HRM分析技术相结合,进行果树重要农艺性状分子标记的开发,是一种行之有效的方法。基于这一策略,本研究鉴定获得了4个与西洋梨矮生性状单显性基因连锁的SNP标记。
梨;矮生性状;;SNP标记;2b-RAD;HRM
0 引言
【研究意义】果树的矮化密植栽培因具有早果、高产、品质优良、易于管理、能够有效节约成本和增加利润等优点,已发展成为国内外极为重要的栽培模式。目前,实现矮密栽培的主要途径是利用矮化砧木和矮生品种,因此,发掘和利用优良的基因资源,进行矮化砧木和矮生品种的培育对果树生产具有重要意义。【前人研究进展】西洋梨矮生型实生变异品种‘Le Nain Vert’具有树体紧凑、矮小的特点,非常适合矮密栽培。研究发现,矮生梨在茎、叶的解剖结构上有区别于普通型梨的显著特征[1]。由于‘Le Nain Vert’的矮生性状受控于显性单基因[2],因此,该基因资源在梨矮生品种的培育方面具有重要应用价值。Wang等[3]将该基因命名为,并通过SSR标记将其定位于西洋梨基因组第16连锁群上。最近,李炜等[4]依据苹果与梨基因组间高度的保守性和共线性关系,以二者的参考基因组序列为基础,又筛选鉴定了一组与连锁的DNA分子标记,将该基因定位到了西洋梨的染色体片段scaffold00074上。利用简化基因组测序技术进行大规模高通量SNP分型是当前国际上动植物基因组学研究热点之一。美国Oregon大学发明的RAD(Restriction Association site DNA)技术是广泛应用的简化基因组测序技术[5]。2007年,Miller等[6]发表了应用芯片杂交的RAD技术。2008年,Baird等[7]将RAD和NGS(Next-generation sequencing)结合,建立了RAD-seq技术。2012年,Peterson等[8]对RAD技术进行改进,提出了ddRAD(double digest RAD)技术。针对RAD技术流程复杂的问题,Elshire等[9]曾提出GBS(genotyping- by-sequencing)技术。随后,Wang等[10]推出基于llB型限制性内切酶的2b-RAD技术,该技术流程简便,重复性和标签代表性高,基于混合泊松分布模型的de novo SNP分型新算法(iML),可使假阳性率得到较大幅度降低[11]。目前,RAD测序技术在许多物种上得到了应用,例如大麦(L. )[12]、黑麦草()[13]等,但在果树上还未见报道。【本研究切入点】为了最终达到分离鉴定的目的,还需要大量的遗传标记进一步对该基因进行更为精细的染色体定位。因此,在以往研究的基础上,继续高效开发更多与其紧密连锁的遗传标记是十分必要的。将2b-RAD测序技术与HRM分析技术相结合,进行果树重要农艺性状SNP标记的开发,是一个值得探索的途径。【拟解决的关键问题】随着西洋梨全基因组序列的公布[14],使大量获得SNP标记成为可能。本研究利用分离群体分组分析的原理[15],采用2b-RAD技术在矮生型/普通型梨对比基因池间进行比较分析,初步筛查可能与目标性状相关的SNP位点,然后利用高效的高分辨率熔解曲线(HRM)分析技术进行大规模的群体分析,以确认与梨矮生性状决定基因共分离的SNP标记,为进一步实现该基因的精细定位及分离鉴定提供重要依据。
1 材料与方法
试验于2016年4—11月在青岛农业大学园艺学院植物遗传改良与育种重点实验室进行。
1.1 基因组DNA 的提取与对比基因池的构建
以‘矮生梨’(L.)ב茌梨’(Rehd.)及‘2-3’(L.×Rehd.)ב绿宝石’(Nakai.)2个F1杂交分离群体(即群体1和群体2)为试材。‘矮生梨’和‘2-3’为矮生型亲本;‘茌梨’和‘绿宝石’为普通型亲本。群体1共有215株,其中矮生型和普通型分别有115株和100株。群体2共有168株,其中矮生型和普通型各有89株和79株。2016年4月取梨树的幼叶,用天根植物基因组DNA提取试剂盒提取并纯化基因组DNA,用琼脂糖凝胶电泳和NanoDrop 2000超微量分光光度计(Thermo Fisher Scientific, USA)对DNA的质量和浓度进行检测,并将其浓度稀释到10 ng·L-1。
从每个群体中分别抽取15个矮生型杂种和15个普通型杂种,将其DNA等量混合,形成矮生型基因池D1和D2以及普通型基因池S1和S2。
1.2 2b-RAD测序分析与SNP位点筛选
利用2b-RAD技术构建梨4个基因池的标签测序文库,并在Hiseq2500 v2平台进行single-end测序(此项工作由上海欧易生物医学科技有限公司完成)。将原始reads进行质量过滤,剔除不含有XⅠ酶切识别位点的序列,剔除低质量序列(即大于10个碱基的质量分数小于20的序列),剔除有10个以上连续相同碱基的序列。从梨参考基因组序列(https:// www.rosaceae.org/species/pyrus/pyrus_communis/genome_v1.0)中提取包含XⅠ酶切位点的标签作为参考序列。将每个样本的高质量reads利用SOAP软件[16](参数设置为:-M4–v2–r0)定位到参考序列上。获得可用于分型的unique标签数目及深度。利用最大似然法(maximum likelihood,ML)进行SNP位点的分型(分型条件:只留取标签内最多有3个SNP的标签位点;分型标签深度设置为500以下。选取在样本间能分型的SNP位点,采用种群结构分析软件对样本间及组间差异SNP位点筛选数据进行分析。
1.3 与连锁的SNP标记的鉴定
对在4个对比基因池上产生的SNP标记进行分析,筛选出矮生型基因池与普通型基因池间的多态性SNP标记,然后查找位于西洋梨染色体片段scaffold00074上的SNP位点,在其两侧合适位置设计引物(Primer 4.0 software),用HRM技术分析这些SNP标记在群体上的分离行为,以确定是否为与紧密连锁的遗传标记。
1.4 高分辨率熔解曲线(HRM)分析
HRM分析在LightCycler®480Ⅱ荧光定量PCR仪(Roche)上进行。反应试剂来自LightCycler®480 High Resolution Melting Master 试剂盒。反应体系为10 μL,内含10 ng基因组DNA 1 μL,1×Master Mix 5 μL,2.0 mmol·L-1MgCl21 μL,上、下游引物(0.2 μmol·L-1)各1 μL,ddH2O 1 μL。PCR扩增程序为95℃预变性10 min,然后按95℃变性10 min、55℃退火15 s、72℃延伸10 s的程序进行45个循环。在PCR循环结束后,立即对扩增产物进行HRM检测,程序为:95℃ 1 min、40℃ 1 min、65℃1 s;在65℃升温至95℃的过程中,以25次/℃的频率收集荧光信息,最后降温至40℃。
HRM分析用LightCycler®480的Gene Scanning软件(1.5 version)进行。
2 结果
2.1 2b-RAD测序结果
对4个样本(即4个近等基因池)的2b-RAD标签测序文库的测序结果共产生67 186 260条reads,平均每个样本测序reads数为16 796 565。将原始reads进行质量过滤后的统计结果表明,每个样本获得平均unique标签数目为86 810,平均测序深度为77×。测序深度能够达到准确分型的标准,可以进行下一步SNP标记分型分析。SOAP软件定位结果表明,4个测序文库中含有酶切位点的高质量reads占测序原始reads的70%以上(表1),表明梨文库的测序质量较好。
2.2 矮生型/普通型对比基因池间SNP位点筛选
选取在样本间能分型的SNP位点,分别在来自群体1和群体2的4个对比基因池间进行比较分析,共筛查出可在矮生型/普通型表型间进行分型的SNP标记1 317个,进一步的标签筛查结果表明,其中有8个SNP位于西洋梨染色体片段scaffold00074(即的定位染色体片段,长度441 741 bp)上,其染色体位置及样品分型结果见表2。
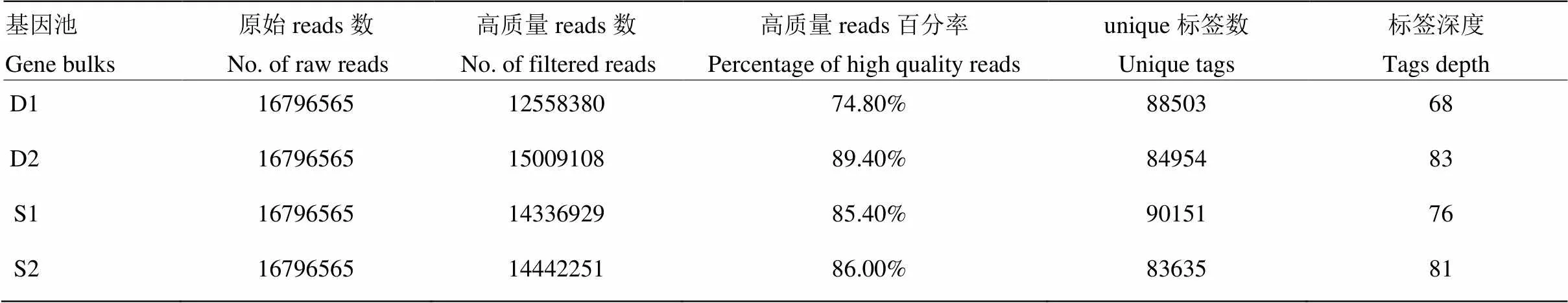
表1 2b-RAD测序结果
D1、D2:分别来自群体1和群体2的矮生型基因池;S1、S2:分别来自群体1和群体2的普通型基因池。下同
D1 and D2 represent the dwarf bulk from population 1 and population 2, respectively. S1 and S2 represent the standard bulk from population 1 and population 2, respectively. The same as below

表2 定位到西洋梨染色体片段scaffold00074上的8个SNP标记及其分型信息
方框为碱基差异位点位置The box represents the position of the base difference
2.3 与位点共分离的SNP标记鉴定
针对以上8个定位于scaffold00074上的SNP位点,分别设计8对PCR引物(表3),并分别在群体1和群体2上进行HRM分析,最终在群体1上检测到SNP1和SNP7两个与位点共分离的SNP标记,在群体2上检测到SNP1、SNP6、SNP7和SNP8 4个可与位点共分离的SNP标记。根据其扩增子的熔解曲线形状差异,可有效地区分矮生型和普通型表型(图1、图2)。在群体1的215个杂种后代和群体2的168个杂种后代中,未发现有标记与性状的重组类型。

表3 8个SNP位点群体分型检测所用的HRM 引物

图 1 SNP1和SNP7在‘矮生梨’ב茌梨’群体中的HRM分析结果

图 2 SNP1、SNP6、SNP7和SNP8在‘2-3’ב绿宝石’群体中的HRM 分析结果
3 讨论
单核苷酸多态性(SNP)是基因组中最常见的遗传多态性。SNP标记已在果树的遗传图谱构建、基因标记与定位等研究中发挥了重要作用[17-20]。与RFLP、AFLP、SSR等传统基因分型技术相比,SNP分子具有分布广泛、代表性强、稳定性好、易于分析等优势。SNP分子标记的获得往往依赖于大规模、高通量的基因组数据分析[21]。对果树来说,其复杂的基因组是造成测序分析成本高的主要原因。
简化基因组测序技术2b-RAD是改进的RAD技术,使测序更加简易、精确,已在高密度遗传图谱的构建、群体的遗传学研究、群体进化分析、标记开发等领域得到了应用[22-28]。高分辨率熔解曲线分析是一种高效的遗传变异检测方法,被广泛的应用于突变扫描、基因分型和标记开发等方面。目前,HRM技术已发展成为SNP标记分型的重要手段[29-31]。李炜等[4]曾依据苹果与梨基因组间高度的保守性和共线性关系,以苹果基因组序列为参考设计了大量引物,通过HRM分析仅筛查到1个与梨矮生性状决定基因紧密连锁的SNP标记。为了提高SNP位点的检测效率,本研究利用2b-RAD测序技术通过对4个对比基因池间共有标签的序列分析,筛查到了大量的可能与目标性状相关的SNP位点,对于初步筛查到的SNP标记,再利用高通量的HRM分析技术进行群体上的大规模验证,以确定其是否为与目标性状紧密连锁的遗传标记。这样,可避免了在群体上继续用2b-RAD技术进行验证分析造成的高成本问题。本研究基于这一策略,鉴定获得了一组与梨矮生性状决定基因位点连锁的新标记。可见,将2b-RAD测序技术与HRM分析技术相结合,进行果树重要农艺性状分子标记的开发,是一种行之有效的方法。
根据不同亲本构建的杂交群体的遗传背景是有差异的,因此,依据不同群体开发的遗传标记是不可能完全相同的。本研究结果显示,在群体1上,鉴定出了2个与连锁的SNP标记;在群体2上,鉴定出了4个与连锁的SNP标记。另外,对在不同群体上鉴定到的相同的SNP标记来说,其HRM分析熔解曲线形状也可能会出现差异。出现这一现象的原因,也与群体的遗传基础差别有关。本研究从2个不同的杂种群体上共检测到了4个新的与梨紧密连锁的DNA分子标记。这些标记,不仅可以作为目标基因定位的参考依据,也可成为目标性状标记辅助选择的有力工具。
4 结论
应用2b-RAD技术对2对矮生型/普通型对比基因池进行基因组测序分析,在对比基因池间筛选出单核苷酸多态性(SNP)位点1 317个,其中有8个SNP位点位于的定位染色体scaffold0007上。用HRM分析技术对这8个SNP位点进行群体上的进一步检测,共鉴定出4个与位点共分离的SNP标记,其中SNP1和SNP7为2对群体共有的SNP标记。这一研究结果对的精细定位及其候选基因的筛查具有重要参考。
References
[1] Chen B Y, Wang C H, Tian Y K, Chu Q, Hu C. Anatomical characteristics of young stems and mature leaves of dwarf pear., 2015, 186: 172-179.
[2] Rivalta L, Dradi M, Rosati C. Thirty years of pear breeding activity at ISF Forli, Italy., 2002, 596(1): 233-238.
[3] Wang C H, Tian Y K, Buck E J, Gardiner S E, Dai H Y, Jia Y. Genetic mapping of PcDw determining pear dwarf trait., 2011, 136(1): 48-53.
[4] 李炜, 田义轲, 王彩虹, 白牡丹, 侯董亮. 通过HRM技术筛查与梨矮生性状决定位点紧密连锁的SNP标记. 园艺学报, 2015, 42(2): 214-220.
Li W, Tian Y K, Wang C H, Bai M D, Hou D L. Screening of SNP markers tightly linked tolocus determining pear dwarf trait using HRM technology., 2015, 42(2): 214-220. (in Chinese)
[5] Davey J W, Blaxter M L. RADSeq: Next-generation population genetics., 2011, 9(5): 416-423.
[6] Miller M R, Dunham J P, Amores A, Cresko W A, Johnson E A. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers., 2007, 17(2): 240-248.
[7] Baird N A, Etter P D, Atwood T S, Currey M C, Shiver A L, Lewis Z A, Selker E U, Cresko W A, Johnson E A. Rapid SNP discovery and genetic mapping using sequenced RAD markers., 2008, 3(10): e3376.
[8] Peterson B K, Weber J N, Kay E H, Fisher H S, Hoekstra H E. Double digest RADseq: An inexpensive method for de novo SNP discovery and genotyping in model and non-model species., 2012, 7(5): e37135.
[9] Elshire R J, Glaubitz J C, Sun Q, Poland J A, Kawamoto K, Buckler E S, Mitchell S E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species., 2011, 6(5): e19379.
[10] Wang S, Meyer E, Mckay J K, Matz M V. 2b-RAD: A simple and flexible method for genome-wide genotyping., 2012, 9(8): 808-810.
[11] Dou J, Zhao X, Fu X, Jiao W, Wang N, Zhang L, Hu X, Wang S, Bao Z. Reference-free SNP calling: improved accuracy by preventing incorrect calls from repetitive genomic regions., 2012, 7(1): 1-9.
[12] Chutimanitsakun Y, Nipper R W, Cuesta-Marcos A, Cistué L, Corey A, Filichkina T, Johnson E A, Hayes P M. Construction and application for QTL analysis of a restriction site associated DNA (RAD) linkage map in barley., 2011, 12(1): 4.
[13] Pfender W F, Saha M C, Johnson E A, Slabaugh M B. Mapping with RAD (restriction-site associated DNA) markers to rapidly identify QTL for stem rust resistance in., 2011, 122(8): 1467-1480.
[14] Chagné D, Crowhurst R N, Pindo M, Thrimawithana A, Deng C, Ireland H, Fiers M, Dzierzon H, Cestaro A, Fontana P, Bianco L, Lu A, Storey R, Knäbell M, Saeedl M, Montanari1 S, Kim Y K, Nicolini D, Larger S, Stefani E, Allan A C, Bowen J, Harvey I, Johnston J, Malnoy M, Troggio M, Perchepied L, Sawyer G, Wiedowl C, Won K, Viola R, Hellens R P, Brewer L, Bus V G , Schaffer R J, Gardiner S E, Velasco R. The draft genome sequence of european pear (L. ‘Bartlett’)., 2014, 9: e92644.
[15] Michelmore R W, Paran I, Kesseli R V. Identification of markers linked to disease-resistance genes by bulked segregant analysis: A rapid method to detect markers in specific genomic regions by using segregating populations., 1991, 88: 9828-9832.
[16] Li R, Yu C, Li Y, Lam T W, Yiu S M, Kristiansen K, Wang J. SOAP2: an improved ultrafast tool for short read alignment., 2009, 25 (15): 1966-1967.
[17] Baumgartner I O, Kellerhals M, Costa F, Dondini L, Pagliarani G, Gregori R, Tartarini S, Leumann L, Laurens F, Patocchi A. Development of SNP-based assays for disease resistance and fruit quality traits in apple (Borkh.) and validation in breeding pilot studies., 2016, 12(3): 1-21.
[18] Zeballos J L, Abidi W, Giménez R, Monforte A J, Moreno M Á, Gogorcena Y. Mapping QTLs associated with fruit quality traits in peach [(L.) Batsch] using SNP maps., 2016, 12(3): 1-17.
[19] Wu J, Li L T, Li M, Khan M A, Li X G, Chen H, Yin H, Zhang S L. High-density genetic linkage map construction and identification of fruit-related QTLs in pear using SNP and SSR markers., 2014, 65(20): 5771-5781.
[20] SUN R, CHANG Y, YANG F, WANG Y, LI H, ZHAO Y, CHEN D, WU T, ZHANG X, HAN Z. A dense SNP genetic map constructed using restriction site-associated DNA sequencing enables detection of QTLs controlling apple fruit quality., 2015, 16(1): 747.
[21] 李全林, 王义发, 韩晴, 袁政, 沈雪芳. 糯玉米‘沪五彩花糯1号’品系特异性2b-RAD分子标记的开发及应用. 植物生理学报, 2016, 52(5): 669-677.
Li Q L, Wang Y F, Han Q, Yuan Z, Shen X F. Development and application of molecular markers for event-specific identification of waxy corn ‘Huwucaihuanuo 1’ based on 2b-RAD technique., 2016, 52(5): 669-677. (in Chinese)
[22] Jiao W, Fu X, Dou J, Li H, Su H, Mao J, Yu Q, Zhang L, Hu X, Huang X, Wang Y, Wang S, Bao Z. High-resolution linkage and quantitative trait locus mapping aided by genome survey sequencing: building up an integrative genomic framework for a bivalve mollusc., 2013, 21(1): 85-101.
[23] Seetharam A S, Stuart G W. Whole genome phylogeny for 21species using predicted 2b-RAD fragments., 2013, 1: e226.
[24] Fletcher R S, Mullen J L, Yoder S, Bauerle W L, Reuning G, Sen S, Meyer E, Juenger T E, McKay J K. Development of a next-generation NIL library infor dissecting complex traits., 2013, 14(1): 655-655.
[25] Guo Y, Yuan H, Fang D, Song L, Liu Y, Liu Y, Wu L, Yu J, Li Z, Xu X. An improved 2b-RAD approach (2b-RAD) offering genotyping tested by a rice (L.) F2 population., 2014, 15 (1): 956.
[26] Li C, Li Y, Bradbury P J, Wu X, Shi Y, Song Y, Zhang D, Rodgers-Melnick E, Buckler E S, Zhang Z. Construction of high-quality recombination maps with low-coverage genomic sequencing for joint linkage analysis in maize., 2015, 13: 78.
[27] Pecoraro C, Babbucci M, Villamor A, Franch R, Papetti C, Leroy B, Ortega-Garcia S, Muir J, Rooker J, Arocha F, Murua H, Zudaire I, Chassot E, Bodin N, Tinti F, Bargelloni L, Cariani A. Methodological assessment of 2b-RAD genotyping technique for population structure inferences in yellowfin tuna ()., 2015, 25: 43-48.
[28] Pauletto M, Carraro L, Babbucci M, Lucchini R, Bargelloni L, Cardazzo B. Extending RAD tag analysis to microbial ecology: A comparison between multilocus sequence typing and 2b-RAD to investigate listeriamonocytogenes genetic structure., 2016, 16(3): 823-835.
[29] Lu Z, Niu L, Chagné D, Cui G, Pan L, Foster T, Zhang R P, Zeng W F, Wang Z Q. Fine mapping of the temperature-sensitive semi-dwarf (Tssd) locus regulating the internode length in peach ()., 2016, 36(2): 1-11.
[30] Wang C H, Li W, Tian Y K, Hou D L, Bai M D. Development of molecular markers for genetic and physical mapping of thelocus in pear (L.)., 2016, 91(3): 299-307.
[31] 赵俊生, 杨晓燕, 曾祥有, 钟声, 方静, 罗剑斌, 曾运友, 向旭. 利用SNP 分子标记分析化橘红种质资源. 分子植物育种, 2016, 14(5): 1203-1211.
Zhao J S, Yang X Y, Zeng X Y, Zhong S, Fang J, Luo J B, Zeng Y Y, Xiang X. Analysis on germplasm resources ofusing SNP molecular markers., 2016, 14(5): 1203-1211.
(责任编辑 赵伶俐)
Development of DNA Molecular Markers for the Dwarf Trait in Pear through the Method of 2b-RAD Sequencing and HRM Analysis
XIAO YuXiong, WANG CaiHong, TIAN YiKe, Yang ShaoLan, Li DingLi, ZHANG HaiYue
(College of Horticulture, Qingdao Agricultural University/Qingdao Key Laboratory of Genetic Improvement and Breeding in Horticultural Plants, Qingdao 266109, Shandong)
【Objective】As an important agronomic trait, the dwarf character of tree is significant to fruit intensive culture. The dwarf traits of pear originated from variety ‘Le Nain Vert’, a chance seedling of, is determined by a dominant gene, which sequence information is still unknown. Development of DNA molecular markers tightly linked to thelocus could provide important information for identifying of this gene.【Method】Using 2 different F1populations obtained from ‘Aishengli’בChili’ and ‘2-3’בLvbaoshi’, respectively, as plant materials, 2 pairs of bulks for dwarf type/standard type were sequenced through IIB restriction association site DNA (2b-RAD) technology, and then single nucleotide polymorphism (SNP) markers were detected between the dwarf and standard bulks based on the principle of bulked segregant analysis. After that, those SNPs located on themapped chromosome were selected and tested on the whole population to confirm their linkage relationship to thegene by high-resolution melting (HRM) analysis.【Result】A total of 67 186 260 reads were produced from the 2b-RAD sequencing of the 4 samples (2 pairs of bulks). That means the average reads per sample was 16 796 565. Statistical analysis of the filtered raw reads showed that the average unique tags per sample were 86 810, and the average sequencing depth was 77× which was enough for accurate genotyping. For the 4 libraries, mapping through SOAP software indicated that the high quality reads containing the restriction site accounted for more than 70% of the raw reads, which means the high quality of sequencing. Totally 1 317 polymorphic SNP markers were screened between the dwarf bulks and standard bulks derived from the 2 different populations, and 8 of them were mapped on the-localized chromosome scaffold00074. Further detection of the 8 SNPs through HRM analysis showed that 2 and 4 SNP markers co-segregated with thelocus were identified from population ‘Aishengli’בChili’ and population ‘2-3’בLvbaoshi’, respectively. According to the different shapes of the melting curves of amplicons, the dwarf/normal phenotype could be distinguished effectively. The recombinants for each marker and the target trait were not found in both of the two populations, which contained 215 and 168 progenies, respectively. 【Conclusion】The combination of 2b-RAD sequencing and HRM analysis technology is an efficient method for exploiting molecular markers of important agronomic traits in fruit trees. In this study, a total of 4 SNP markers co-segregated with thelocus were identified based on this strategy.
pear; dwarf traits;; SNP marker; 2b-RAD; HRM
2017-01-20;接受日期:2017-04-13
国家自然科学基金(31372049)、山东省现代农业产业技术体系果品创新团队(SDAIT-06-06)
肖玉雄,E-mail:1427449415@qq.com。通信作者王彩虹,E-mail:chwang@qau.edu.cn
猜你喜欢
现代仪器与医疗(2021年4期)2021-11-05
今日农业(2021年11期)2021-08-13
煤气与热力(2021年5期)2021-07-22
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
中成药(2018年7期)2018-08-04
中华骨与关节外科杂志(2017年1期)2017-05-17
制造技术与机床(2015年10期)2015-04-09
中国卫生标准管理(2015年14期)2015-01-27
中国中医药现代远程教育(2014年21期)2014-03-01
