
配体稳定的全主族金属芳香性三明治理论研究
2017-09-07 06:23:47尤雪瑞翟华金
山西大学学报(自然科学版) 2017年3期
尤雪瑞,翟华金
(山西大学 分子科学研究所 纳米团簇实验室,山西 太原 030006)
配体稳定的全主族金属芳香性三明治理论研究
尤雪瑞,翟华金*
(山西大学 分子科学研究所 纳米团簇实验室,山西 太原 030006)
文章对配体稳定的合成相主族金属配合物Al[Al[N(SiMe3)2]]6-(1)进行模型简化和成键分析,由此提出全主族金属芳香性三明治概念。采用密度泛函理论在PBE0/def2-TZVP水平上对简化的模型金属配合物团簇[Al[Al(NH2)]6]-(2)作结构优化及频率计算,并利用正则分子轨道(CMO)、适应性自然密度划分(AdNDP)、轨道成分分析等系列手段探究体系的成键特征。基于分子轨道分析团簇2的零级成键图像为[Al(NH2)]32-Al3+[Al(NH2)]32-,轨道成分分析则给出完备成键图像[Al(NH2)]30.35-Al0.30-[Al(NH2)]30.35-,后者与自然键轨道(NBO)结果类似。体系含近似7中心2电子离域π和σ键各一个,同时其核独立化学位移(NICS)为负值,表明团簇2具有π和σ双重芳香性。体系中两个[Al(NH2)]3金属层与Al夹心之间发生复杂的电荷反馈与负反馈,将多至4.5个电子从上下金属层和夹心转移到金属层-夹心之间的6个Al-Al棱上,由此形成和稳定全金属三明治结构。电荷转移过程可利用轨道成分分析进行定量的跟踪。
全主族金属芳香性三明治;纳米团簇;合成配合物;新型化学键;密度泛函理论
0 引言
以20世纪50年代初合成的二茂铁[1]为代表,“三明治化合物”或“夹心化合物”至今已有60余年持续研究历程。典型三明治化合物是指由金属原子和两个环多烯形成的夹心式化合物。环多烯含离域π键,能作为π电子给予体与金属原子形成配位化合物。二茂铁作为首例具有芳香族性质的有机过渡金属三明治化合物,其发现展开了环戊二烯基与过渡金属π配合物的丰富化学,也为有机金属化学研究掀开新的帷幕[2-3]。进入21世纪以来,三明治化合物领域取得两项里程碑式的进展。其一,美国科学家2002年合成一种全新的“不含碳”三明治化合物[P5TiP5]2-,利用无机芳香性配体P5-代替芳香族环多烯[4]。其二,日本研究团队2006年将单原子金属夹心拓展为多原子金属层(但配体仍为环多烯)[5-6]。最近,作者与中科院合作者通过合成实验与量子化学理论计算[7-8]表征了首例全金属芳香性三明治化合物[Sb3Au3Sb3]3-。该化合物由一个夹心Au3金属层和两层全金属芳香性Sb3环状配体相互叠合而成,总体呈三棱柱结构,具有优美的D3h对称性。从学科发展角度,全金属芳香性三明治化合物属该领域的一项重要进展,将进一步激发人们对于新型三明治化合物的兴趣与探索。
理论上,化学家们预测了一系列以第13/14/15主族元素团簇(包括金属团簇)为配体、过渡金属原子为夹心的三明治化合物,例如[Al4TiAl4]2-等[9-10]。这些体系通常只能被视为模型化合物,因为对其势能面的了解远非充分。那么,是否可能用主族金属原子作为夹心、以主族金属团簇作为配体,形成全主族金属三明治化合物呢?如果可能,全主族金属三明治是否具有芳香性?全主族金属三明治化合物能否以合成相形式存在?本文研究结果对上述问题作出肯定的回答。1999年德国Schnöckel等[11]合成了一种类三明治结构Al[Al[N(SiMe3)2]]6-(1)晶体化合物,其中全主族金属夹心结构[Al3-Al-Al3]-作为化合物的核心,在[N(SiMe3)2]6配体作用下稳定存在。但文献中一直未见对此化合物的理论计算和成键分析。
在过去十余年里,我们一直致力于研究气相团簇[12-20]和合成化合物团簇[7-8,21-22]的结构、电子特性和新颖成键。就自由团簇而言我们的研究主题涉及硼球烯(全硼富勒烯)[12]、平面硼团簇[13-14]、硼羰基化学[16,18]、过渡金属d-轨道芳香性、δ-芳香性[15]、全金属芳香三明治[8]、结构流变团簇[17]等物理化学前沿方向。在本项工作中我们尝试从理论层面阐释主族金属夹心化合物Al[Al[N(SiMe3)2]]6-(1)的结构和成键本质。为此我们设计了更为简化的模型团簇[Al[Al(NH2)]6]-(2),即以H原子取代合成相中的SiMe3基团。注意到从与Al3环配位的角度讲,H和SiMe3都是一价σ配体,因此模型团簇2可合理地反映合成相配合物1的结构和成键特性。我们利用密度泛函理论(DFT)[23]对模型团簇2进行结构优化和频率计算,通过正则分子轨道(CMO)、适应性自然密度划分(AdNDP)[24]、核独立化学位移(NICS)[25]等分析揭示该体系本质上具有独特的π和σ双重芳香性。自然键轨道(NBO)[26]和轨道成分分析定量化地展示金属层和夹心之间的电子反馈与负反馈过程。基于本工作,我们确立全主族金属芳香性三明治化合物的概念。
1 理论方法
密度泛函理论[23]由Hohenberg、Kohn和Sham等共同建立并逐渐发展成为电子结构计算的首选方法。本工作使用的PBE0方法[27]在PBE基础上引入25%的HF交换项,掺入的成分是通过理论推导而来,不含拟合参数,因此保持了从头算方法的很多优点,又较为节省计算时间。我们应用Gaussian 09[28]程序包,在PBE0/def2-TZVP水平上对[Al[Al(NH2)]6]-(2)的几何结构进行全优化,并进行振动频率计算,结果表明所得优化几何结构对应势能面上的能量极小点(无虚振动频率)。正则分子轨道和适应性自然密度划分[24]用于分析体系的成键特性,其结果借助Molekel程序[29]实现可视化。自然键轨道[26]分析用于获得自然电荷布居分布(NPA)和韦伯键级(WBI)。核独立化学位移[25]计算用来辅助表征体系的芳香性。轨道成分分析借助Multiwfn[30]程序完成。计算中所有收敛精度均取程序设定的默认值。
2 结果与讨论
2.1 模型团簇D3d[Al[Al(NH2)]6]-(2):优化结构、韦伯键级、自然电荷分布
我们从Schnöckel等报道的合成相晶体结构[11]获得S6点群Al[Al[N(SiMe3)2]]6-(1)初始结构(图1(a))。其中(SiMe3)2配体较为庞大,但与N原子相互作用仅为经典两中心σ单键。为此我们用12个H原子代替12个SiMe3基团,设计出简化模型团簇。在PBE0/def2-TZVP水平对其进行优化和频率计算,得到D3d[Al[Al(NH2)]6]-(2)结构且无虚频,如图1(b)所示。
团簇2可视为Al7内核与外围6个NH2配体相互作用形成的配合物。其中Al7内核由上下两个Al3正三角形金属环与中心Al原子经由6个层间Al—Al链接结合而成,呈三明治构型。团簇2的理论优化结构参数(键长、键角)及其与合成相配合物1实验结果的比较见表1。为便于区分,7个Al原子标注为两类:两个Al3环上的6个Al原子记作Al(i);夹心Al原子记作Al(ii)。可以发现:团簇2的计算键长Al(i)—Al(i)、Al(i)—Al(ii)、Al(i)—N分别为2.53、2.74、1.81 Å,与晶体1实验结果十分吻合。此外计算所得团簇2的∠Al(i)Al(i)Al(i)和∠Al(i)Al(ii)Al(i)键角也与晶体1严格一致,而∠Al(ii)Al(i)N键角则有所差别(团簇2∶132.3°;晶体1∶138.00°);这一差异主要源于晶体1中尺寸较大的SiMe3配体所导致的空间位阻效应。上述结构信息表明团簇2是晶体1的可信模型,同时表明PBE0/def2-TZVP理论水平对本体系的适用性。

Fig.1 (a) Original S6 Al[Al[N(SiMe3)2]]6-(1) complex based on reference [11] and(b) optimized structure of a model D3d[Al[Al(NH2)]6]-(2) cluster at PBE0/def2-TZVP level.Cluster 2 is constructed from complex 1 via isovalent substitution of the SiMe3groups by H图1 (a) 原始S6点群Al[Al[N(SiMe3)2]]6-(1)合成配合物,(b) 在PBE0/def2-TZVP水平下优化的模型D3d[Al[Al(NH2)]6]-(2)团簇。团簇2由配合物1经配体等电子替代实现,即用12个H原子替代12个SiMe3基团
李泽生等[31]理论研究Al3+/0/-得到Al—Al键长为2.54 Å;Martinez[32]研究D3hAl3-得到键长为2.52 Å。因此,结合Pyykkö[33]推荐的Al—Al共价单键键长(2.52 Å),我们认定团簇2中6个Al(i)—Al(i)链接可视为单键;而层间Al(i)—Al(ii)链接则显著伸长,弱于单键,即Al(ii)与Al(i)之间没有定域键。类似地,外围Al(i)—N和N—H链接可近似看作单键。注意到Al(i)—N键长明显短于推荐的单键键长[33],暗示Al(i)—N可能具有一定的多重键特征。韦伯键级支持上述大部分结论(表1)。需要注意的是Al(i)—N的韦伯键级仅为0.52,与其短键长似有矛盾。主要原因是Al(i)—N键兼具较强的共价键与离子键特征。实际上NH2配体总的自然电荷为-0.68 |e|,而Al(i)为+0.59 |e|。众所周知,韦伯键级通常不能准确反映离子键的贡献。
2.2 成键特征:正则分子轨道分析、零级图像、π和σ双重芳香性
Al、N、H的电子组态分别为3s23p1,2s22p3和1s1,加上额外电荷,团簇2共有64个价电子,其32个占据价轨道见图2-图4。如2.1节所述,团簇2外围6个N—Al和12个N—H键可看作σ单键,此外N原子存在6个电子孤对,上述外围Lewis成键元素共消耗48个电子,分子轨道如图2所示。这些轨道比较容易理解。以图2(a)为例,HOMO-5/HOMO-7为两个N3“环”上N 2pz完全成键轨道的正相与反相叠加,它们可还原为两个3中心2电子(3c-2e)轨道,每个N3环一个。同理,HOMO-4/HOMO-6′和HOMO-4′/HOMO-6可组合为2对简并3c-2e轨道,每个N3环一对。这样每个N3环含一个完全成键轨道和一对简并的、部分成键/反键轨道,其进一步组合即为三个N 2pz孤对。简而言之,图2(a)显示为六对N 2pz孤对。类似地,图2(b)和2(c)分别为6个N—Al和12个N—H定域2c-2e σ单键。

表1 结构1和2的键长、键角,以及自然键轨道分析所得的键级和自然电荷
扣除上述外围Lewis成键元素,团簇2中Al7内核可视为16电子体系。其中12个电子主要参与上下层Al3环的Al—Al σ单键,其对应轨道如图3所示。这组轨道与图2(a)或2(b)存在形式上的对应关系,其定域性不难确定和理解。至此体系仅剩余4个电子,它们对于体系的成键至关重要,其分子轨道如图4所示。HOMO和HOMO-3均为离域轨道,主要集中于Al7核上(含向Al-NH2单元的延伸),具有近似7c-2e特征。表2列出它们的详细轨道成分[30]。可以发现,夹心Al(ii)原子对这二个轨道的贡献仅为21%~23%,居于次要地位;而在HOMO中上下Al3环各贡献约31%,在HOMO-3中上下[Al(NH2)]3环各贡献约37% (其中Al3占14%,N3占23%)。这一方面表明团簇2中额外电子主要分布在上下Al3环,另一方面说明上下层Al3或[Al(NH2)]3环主导体系的离域成键。基于轨道分析有两点结论:(1)团簇2中找不到任何一个由夹心Al(ii)原子占主导地位的分子轨道,夹心Al(ii)形式上处于Al3+价态;(2)作为“零级近似图像”,团簇2可表述为[Al(NH2)]32-Al3+[Al(NH2)]32-。

Fig.2 Pictures of CMOs of 2 associated with the N—Al and N—H peripheral bonding:(a) six N 2p lone-pairs,(b) six N—Al σ single bonds,(c) twelve N—H σ single bonds图2 团簇2中Al7的外围成键:(a) 6个N 2p孤对电子,(b) 6个N—Al σ单键,(c) 12个N—H σ单键
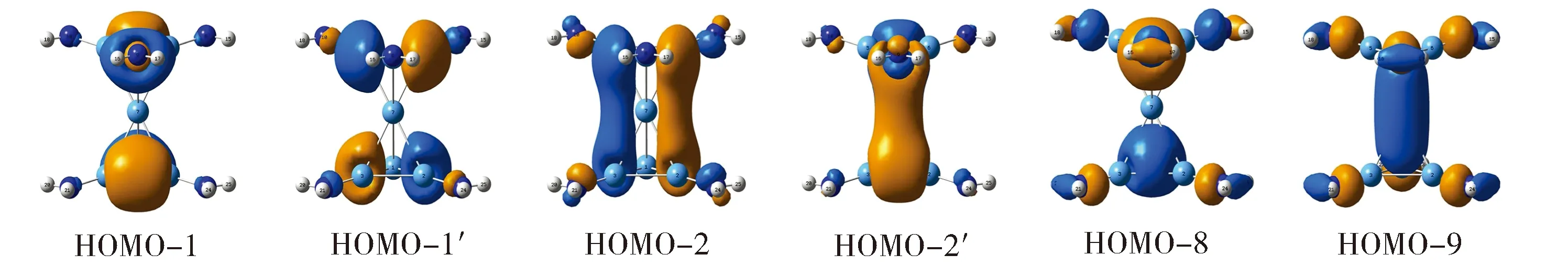
Fig.3 Pictures of CMOs of 2 that are responsible for Al—Al σ bonds within the Al3 rings图3 团簇2上下层Al3环6个Al—Al σ单键对应的分子轨道
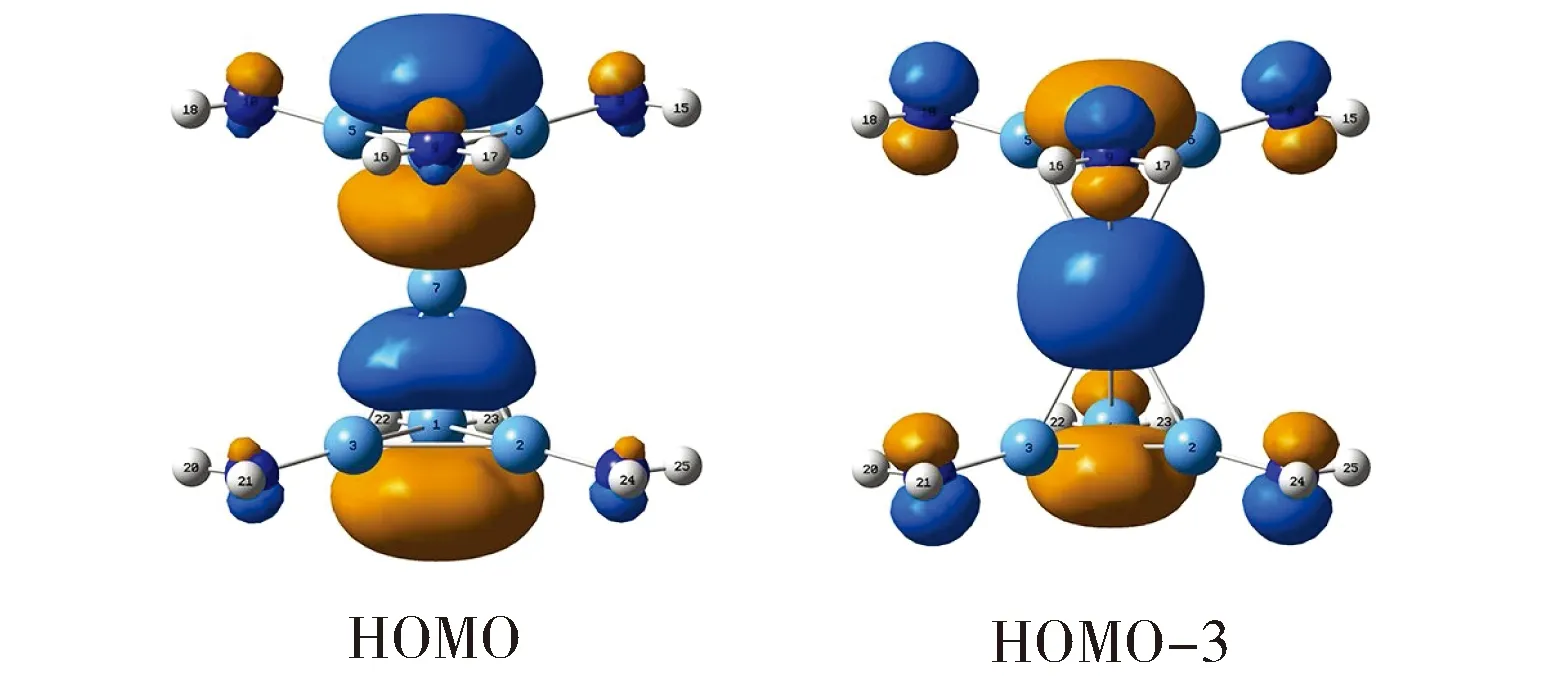
Fig.4 Pictures of two delocalized 7c-2e CMOs in cluster 2图4 团簇2中的两个离域7c-2e键HOMO和HOMO-3分别为π和σ轨道
具体分析到离域轨道的性质,HOMO-3中上下N3“环”的成分形式上具有π特征,但因N…N间距较大,只能近似为对N孤对的部分贡献;上下Al3环的主要贡献为s/p杂化,具有σ特征,表明体系具有一定程度的σ芳香性。必须指出,σ芳香性总体上是微弱的,因为每个Al3对HOMO-3的贡献仅为14% (表1),即:团簇2中Al3环的σ芳香性近似为典型3c-2e σ芳香体系的1/7。HOMO则为Al3环占主导的离域π轨道,两个Al3环各贡献31%,赋予体系较强的π芳香性。可以认为,Al3环π芳香性近似为典型3c-2e π芳香体系的1/3。总而言之,分子轨道分析表明团簇2含离域π和σ轨道各一个,它们赋予上下Al3环π和σ双重芳香性;其中π芳香性较强,而σ芳香性则相对微弱。
值得特别指出的是,三明治团簇2中各[Al(NH2)]32-芳香环无须分别满足(4n+2) Hückel规则,而可以通过两个芳香环之间的耦合集体上实现电子计数,即各芳香环仅需提供、甚至只是形式上提供Hückel规则要求电子数的一半。因此,每个[Al(NH2)]32-芳香环虽然只有离域π和σ电子各一个,却能成就团簇2的2π和2σ双重芳香性 (图4)。
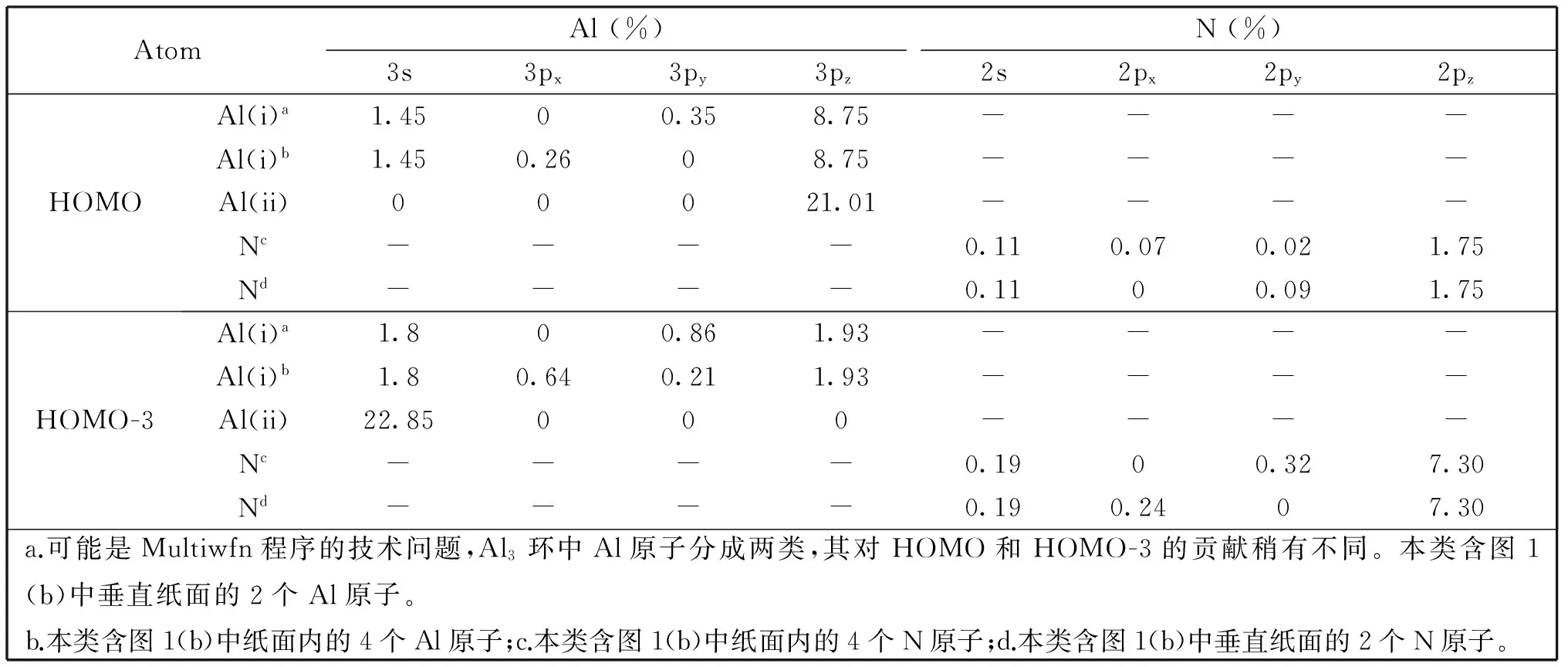
表2 团簇2中HOMO和HOMO-3的轨道成分分析
2.3 成键特征:AdNDP分析和NICS计算
上节的轨道分析结果可用AdNDP[24]数据更直观地进行表达。AdNDP方法是NBO分析的一种拓展,它将分子体系的成键描述为n中心2电子(nc-2e)键,其中n的取值从1直至体系所含的总原子数。因此AdNDP不仅能还原经典Lewis成键元素(孤对和2c-2e定域键),也能揭示体系的非经典多中心成键(芳香性)。图4为团簇2的AdNDP方案:第一行展示6个外围N 2p孤对、6个N-Al σ定域单键、12个N-H σ定域单键。第二行则为Al7核中的6个环内Al—Al σ定域单键和2个离域键(π和σ各一个)。这一成键图像与上节完全一致,再次表明团簇2具有双重芳香性。有趣的是Al3环内Al—Al 2c-2e σ键占据数较低(1.63 |e|),这与Al向N的电荷转移有关(见表1);离域π和σ键虽然对环内Al—Al键贡献微弱(表2),但足以补偿之,使其键长达到单键水平。

Fig.5 AdNDP bonding pattern for cluster 2.Occupation numbers (ONs) are shown轨道占据数(ON)已列出图5 团簇2的AdNDP成键图像
为了辅助表征团簇2体系的芳香性,我们计算其NICS[25]值,因为它具有简便性和高效性,可作为判断芳香性的一种独立的、强有力的证据。在PBE0/def2-TZVP水平下计算Al3环中心及环上1 Å处的NICS和NICSzz值,结果见表3。所有NICS(0)值均为负,显示体系具有σ芳香性;NICS(1)值为负则说明体系具有π芳香性。
2.4 三明治结构的形成:电子反馈与负反馈

表3 团簇2的NICS计算结果。R为计算点距离Al3环中心的距离
前面2.2和2.3节的成键分析揭示,团簇2是一个配体稳定的全主族金属芳香性三明治。全金属三明治的层间Al(i)—Al(ii)成键如何形成?零级图像[Al(NH2)]32-Al3+[Al(NH2)]32-与NBO电荷分布[Al(NH2)]30.28-Al0.44-[Al(NH2)]30.28-(表1)之间的差异如何协调?为厘清上述问题,我们通过轨道成分分析研究上中下三层之间的电荷转移过程。
仔细观察轨道图像(图2-图4)不难看出,虽然每个轨道都可以近似地指定为孤对/单键/离域键,但多个轨道含有我们尚未讨论的“次级成分”。比如,HOMO-2/HOMO-2′主要归于上下Al3环内Al(i)—Al(i) 2c-2e σ键,但它们却包含显著的Al(ii) 2p成分(26%)。又如,HOMO-7属N 2p孤对电子,却有Al(ii) 2s参与(7.5%)。类似这样的轨道可列出11个,其次级成分均超过5%,见表4。表中第一行假定体系的额外电荷分布在上下[Al(NH2)]3层,而夹心Al(ii)亦向上下层负反馈所有价电子,呈Al(ii)3+价态,此即前面2.2节的零级图像。第二、三行修正离域轨道HOMO和HOMO-3中Al(ii)的成分。上述二项的综合效应是夹心Al(ii)原子向上下金属层负反馈2.12 |e| (未计额外电荷)。后面9个轨道反映上下层芳香环向夹心Al(ii)的电子反馈,共计2.42|e|。上述11个轨道的次级成分展示强大的集体效应,使零级图像的Al(ii)3+价夹心原子回复到Al(ii)0.30-,这十分接近NBO电荷态Al0.44-(表1)。作为一种极其粗略的近似,我们假定从上下金属层向夹心反馈的电子(2.42 |e|)和由夹心向上下金属层负反馈的电子(2.12 |e|)累积在层间的6个Al(i)—Al(ii)链接上,可给出其键级为~0.4,这与计算所得韦伯键级(0.61;表1)可比拟。我们指出,电子反馈、负反馈是共价作用的典型表现形式,使上下金属层和夹心金属原子相互粘合形成三明治结构,而上下金属环的π和σ双重芳香性则进一步稳定该全金属三明治体系。
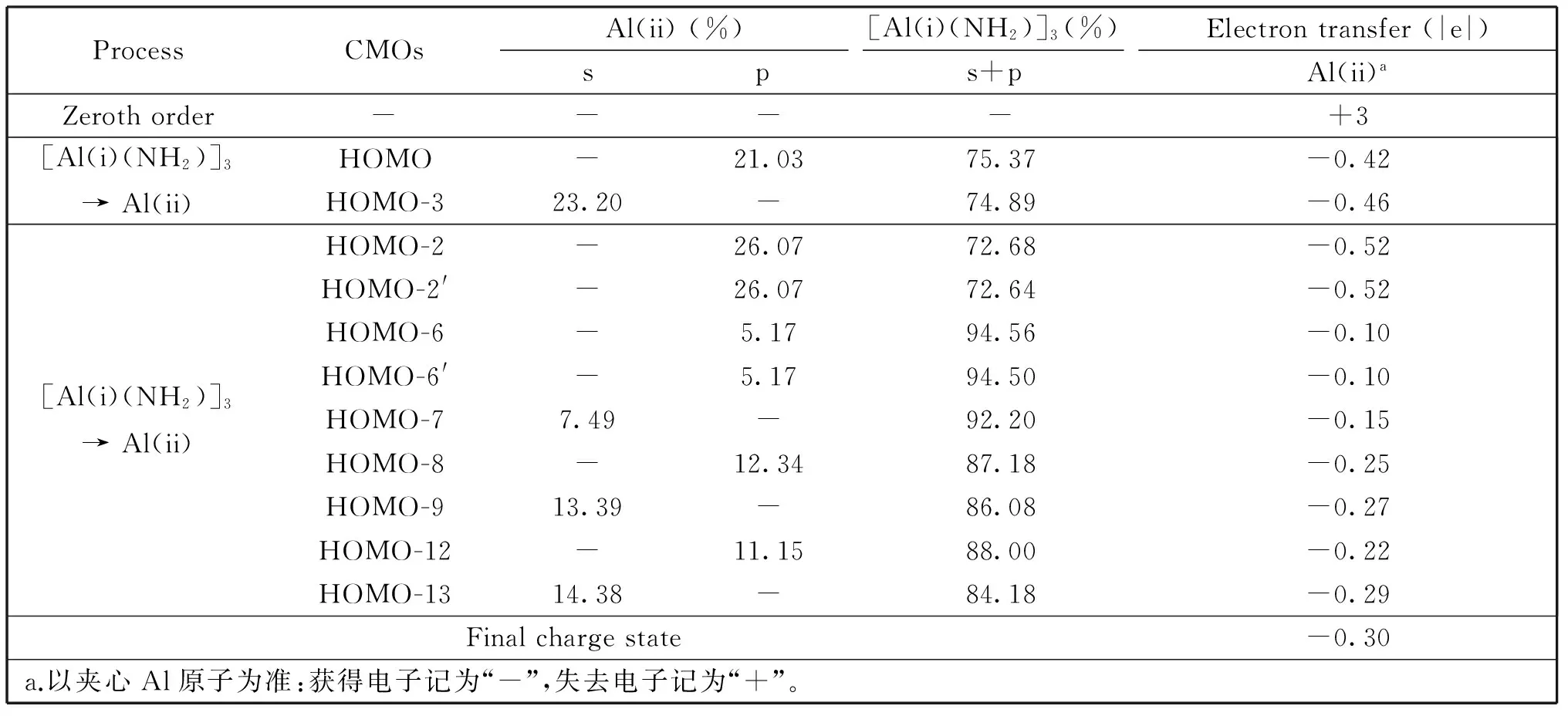
表4 团簇2中的电荷转移过程
3 结论
在密度泛函理论水平下研究了模型三明治团簇[Al[Al(NH2)]6]-的结构、电子特性及成键。该团簇的上下Al3(NH2)3环呈交错构型,夹裹一个Al中心,整体具有D3d对称性,与文献报道的一种合成相配合物类似。正则分子轨道、AdNDP、NBO、NICS等分析表明模型团簇含离域π和σ键各一个,具有双重芳香性。体系的零级近似图像为[Al(NH2)]32-Al3+[Al(NH2)]32-,而NBO有效图像则为[Al(NH2)]30.28-Al0.44-[Al(NH2)]30.28-。详细的轨道成分分析跟踪电荷在金属层和金属夹心之间的反馈与负反馈过程,可定量地解释零级图像与有效成键图像之间的差异。电子反馈与负反馈稳定三明治体系,并可粗略解释层间金属-金属链接的键级。本工作确立配体稳定的全主族金属芳香性三明治化合物的科学概念。
[1] Wilkinson G,Rosenblum M,Whiting M C,etal.The Structure of Iron Bis-Cyclopentadienyl[J].JAmChemSoc,1952,74:2125-2126.DOI:10.1021/ja01128a527.
[2] Streitwieser A Jr,Müller-Westerhoff U.Bis(cyclooctatetraenyl)uranium (Uranocene).A New Class of Sandwich Complexes that Utilize Atomic f Orbitals[J].JAmChemSoc,1968,90:7364.DOI:10.1021/ja01028a044.
[3] Vidyaratne I,Nikiforov G B,Gorelsky S I,etal.Isolation of a Self-Activating Ethylene Trimerization Catalyst[J].AngewChemIntEd,2009,48:6552-6556.DOI:10.1002/anie.200900957.
[5] Murahashi T,Fujimoto M,Oka M,etal.Discrete Sandwich Compounds of Monolayer Palladium Sheets[J].Science,2006,313:1104-1107.DOI:10.1126/science.1125245.
[6] Murahashi T,Kato N,Uemura T,etal.Rearrangement of a Pd4Skeleton from a 1D Chain to a 2D Sheet on the Face of a Perylene or Fluoranthene Ligand Caused by Exchange of the Binder Molecule[J].AngewChemIntEd,2007,46:3509-3512.DOI:10.1002/anie.200700340.
[7] Pan F X,Li L J,Wang Y J,etal.An All-Metal Aromatic Sandwich Complex[Sb3Au3Sb3]3-[J].JAmChemSoc,2015,137:10954-10957.DOI:10.1021/jacs.5b07730.
[8] You X R,Tian W J,Li D Z,etal.On the Nature of Chemical Bonding in the All-Metal Aromatic[Sb3Au3Sb3]3-Sandwich Complex[J].PhysChemChemPhys,2016,18:13423-13431.DOI:10.1039/c6cp00101g.
[9] Mercero J M,Ugalde J M.Sandwich-Like Complexes Based on “All-Metal” (Al42-) Aromatic Compounds[J].JAmChemSoc,2004,126:3380-3381.DOI:10.1021/ja039074b.
[10] Yang L M,Ding Y H,Sun C C.Sandwich-like Compounds Based on the All-Metal Aromatic Unit Al42-and the Main-Group Metals M (M=Li,Na,K,Be,Mg,Ca)[J].ChemEurJ,2007,13:2546-2555.DOI:10.1002/chem.200601223.
[11] Purath A,Köppe R,Schnöckel H.[Al7{N(SiMe3)2}6]-:A First Step Towards Aluminum Metal Formation by Disproportionation[J].AngewChemIntEd,1999,38:2926-2928.
[12] Zhai H J,Zhao Y F,Li W L,etal.Observation of an All-Boron Fullerene[J].NatChem,2014,6:727-731.DOI:10.1038/NCHEM.1999.
[13] Zhai H J,Kiran B,Li J,etal.Hydrocarbon Analogues of Boron Clusters-Planarity,Aromaticity and Antiaromaticity[J].NatMater,2003,2:827-833.DOI:10.1038/nmat1012.
[14] Zhai H J,Alexandrova A N,Birch K A,etal.Hepta-and Octacoordinate Boron in Molecular Wheels of Eight-and Nine-Atom Boron Clusters[J].AngewChemIntEd,2003,42:6004-6008.DOI:10.1002/anie.200351874.
[15] Zhai H J,Averkiev B B,Zubarev D Y,etal.δ Aromaticity in[Ta3O3]-[J].AngewChemIntEd,2007,46:4277-4280.DOI:10.1002/anie.200700442.
[16] Zhai H J,Chen Q,Bai H,etal.Boronyl Chemistry:The BO Group as a New Ligand in Gas-Phase Clusters and Synthetic Compounds[J].AccChemRes,2014,47:2435-2445.DOI:10.1021/ar500136j.
[17] Wang Y J,Zhao X Y,Zhai H J,etal.B11-:A Moving Subnanoscale Tank Tread[J].Nanoscale,2015,7:16054-16060.DOI:10.1039/c5nr03732h.
[18] Zhai H J,Li S D,Wang L S.Boronyls as Key Structural Units in Boron Oxide Clusters:B(BO)2-and B(BO)3-[J].JAmChemSoc,2007,129:9254-9255.DOI:10.1021/ja072611y.
[21] Popov I A,Pan F X,You X R,etal.Peculiar All-Metal σ-Aromaticity of the[Au2Sb16]4-Anion in the Solid State[J].AngewChemIntEd,2016,55:15344-15346.DOI:10.1002/anie.201609497.
[22] You X R,Feng L Y,Li R,etal.Chemical Bonding and σ-Aromaticity in Charged Molecular Alloys:[Pd2As14]4-and[Au2Sb14]4-Clusters[J].SciRep,2017,7:791.DOI:10.1038/s41598-017-00867-5.
[23] Hohenberg P,Kohn W.Inhomogeneous Electron Gas[J].PhysRev,1964,136:864-871.DOI:10.1103/PhysRev.136.B864.[24] Zubarev D Y,Boldyrev A I.Developing Paradigms of Chemical Bonding:Adaptive Natural Density Partitioning[J].PhysChemChemPhys,2008,10:5207-5217.DOI:10.1039/b804083d.
[25] Schleyer P v R,Maerker C,Dransfeld A,etal.Nucleus-Independent Chemical Shifts:A Simple and Efficient Aromaticity Probe[J].JAmChemSoc,1996,118:6317-6318.DOI:10.1021/ja960582d.
[26] Glendening E D,Badenhoop J K,Reed A E,etal.NBO 5.0[CP],Theoretical Chemistry Institute,University of Wisconsin,Madison (2001).
[27] Adamo C,Barone V.Toward Reliable Density Functional Methods without Adjustable Parameters:The PBE0 Model[J].JChemPhys,1999,110:6158-6170.DOI:10.1063/1.478522.
[28] Frisch M J Trucks G W,Schlegel H B,etal.GAUSSIAN 09,Revision D.01[CP].Gaussian,Inc.,Wallingford,CT (2009).[29] Varetto U.Molekel 5.4.0.8[CP].Swiss National Supercomputing Center,Manno,Switzerland (2009).
[30] Lu T,Chen F W.Calculation of Molecular Orbital Composition[J].ActaChimSinica,2011,69:2393-2406.
[31] Sun J,Li Z S,Sun C C,etal.Theoretical Study of Alnand AlnO (n=2-10) Clusters[J].JPhysChemA,2006,110:2729-2738.DOI:10.1021/jp051033+.
[32] Alcantar-Medina K O,Herrera-Trejo M,Martinez A I,etal.Evolution of the Structural and Electronic Properties of Small Alkali Metal-Doped Aluminum Clusters[J].ComputTheorChem,2017,1099:55-63.DOI:10.1016/j.comptc.2016.11.008.
[33] Pyykkö P.Additive Covalent Radii for Single-,Double-,and Triple-Bonded Molecules and Tetrahedrally Bonded Crystals:A Summary[J].JPhysChemA,2015,119:2326-2337.DOI:10.1021/jp5065819.
Ligand-Stabilized All-Main-Group-Metal Sandwich Complex:Structure,Bonding,and Aromaticity
YOU Xuerui,ZHAI Huajin*
(Nanocluster Laboratory,Institute of Molecular Science,Shanxi University,Taiyuan 030006,China)
We model,at the density-functional theory (DFT) level,a synthetic main group metal complex using the[Al[Al(NH2)]6]-cluster and propose a concept of ligand-stabilized all-main-group-metal aromatic sandwich.The structural optimization and frequency calculations at PBE0/def2-TZVP indicate thatD3d[Al[Al(NH2)]6]-cluster is a true minimum.Canonical molecular orbital (CMO) and adaptive natural density partitioning (AdNDP) analyses suggest that at the zeroth order the cluster can be formulated as[Al(NH2)]32-Al3+[Al(NH2)]32-.A more realistic model is[Al(NH2)]30.35-Al0.30-[Al(NH2)]30.35-,via quantitative orbital composition analysis,and natural bond orbital (NBO) data give a similar picture.The CMO and AdNDP analyses reveal a delocalized 7-center 2-electron (7c-2e) π bond,as well as a 7c-2e σ bond,rendering the system π and σ double aromaticity,which is also supported by nucleus independent chemical shift (NICS) calculations.Electron donation and back-donation occur in between two[Al(NH2)]3layers and the Al core,collectively accumulating as many as 4.5 electrons along the interlayer Al—Al edges,which stabilize the all-metal sandwich.
all-main-group-metal aromatic sandwich;nanoclusters;synthetic compounds;chemical bonding;density-functional theory
10.13451/j.cnki.shanxi.univ(nat.sci.).2017.03.019
2017-06-15;
2017-06-20
国家自然科学基金 (21573138);山西省“三晋学者”支持计划
尤雪瑞(1992-),女,山西吕梁人,博士研究生,研究领域为理论与计算化学。
翟华金(ZHAI Huajin),E-mail:hj.zhai@sxu.edu.cn
O641
A
0253-2395(2017)03-0540-08
猜你喜欢
高中数理化(2023年6期)2023-08-26 13:28:24
辽宁科技大学学报(2022年5期)2023-01-04 12:45:34
大学化学(2021年8期)2021-09-26 10:50:46
山东化工(2020年5期)2020-04-07 09:59:30
原子与分子物理学报(2020年5期)2020-03-17 06:59:34
物理化学学报(2019年2期)2019-03-08 08:30:36
数理化解题研究(2018年28期)2018-11-08 02:30:58
考试周刊(2018年39期)2018-04-19 10:39:44
求学·理科版(2016年3期)2016-03-23 05:46:24
中学生数理化·高一版(2015年1期)2015-07-30 13:05:09
