
荧光标记LDR⁃PCR复合扩增方法对高度降解DNA检材的SNPs分型研究
2017-09-03 10:01邢佳鑫孙溢华宣金锋姚军丁梅庞灏李春梅夏皙王保捷
中国医科大学学报 2017年8期
邢佳鑫,孙溢华,2,宣金锋,姚军,丁梅,庞灏,李春梅,夏皙,王保捷
(1.中国医科大学法医学院法医血清教研室,沈阳 110122;2.苏州市公安局吴中分局法医DNA室,江苏 苏州 215104)
荧光标记LDR⁃PCR复合扩增方法对高度降解DNA检材的SNPs分型研究
邢佳鑫1,孙溢华1,2,宣金锋1,姚军1,丁梅1,庞灏1,李春梅1,夏皙1,王保捷1
(1.中国医科大学法医学院法医血清教研室,沈阳 110122;2.苏州市公安局吴中分局法医DNA室,江苏 苏州 215104)
目的构建一套基于连接酶检测反应(LDR)的荧光标记PCR复合扩增体系,为解决高度降解DNA法医学分析提供新的策略。方法选择8个单核苷酸多态性(SNPs)位点(rs10802248、rs10516197、rs10488372、rs2278945、rs4757318、rs4887255、rs4889002和rs9304473),设计合成各SNPs位点的LDR探针和连接产物PCR引物,通过LDR将连接产物进行PCR扩增,并将扩增产物进行毛细管凝胶电泳,构建复合扩增SNPs分型体系。结果使用荧光标记LDR-PCR复合扩增方法,对不同个体的8个SNPs位点进行分型,分型结果与测序结果完全一致;对于甲醛固定石蜡包埋组织(FFPET)检材高度降解的DNA样品,荧光标记LDR-PCR复合扩增体系能够实现8个SNPs位点的准确分型。结论荧光标记LDR-PCR复合扩增方法能够对8个SNPs位点进行一次性分型,结果准确、可靠,是一种简单、高效并且较为实用的SNPs分型新方法,适用于高度降解检材的检测。
法医物证学;连接酶检测反应;复合扩增;降解DNA;单核苷酸多态性
在实际检验过程中,法医鉴定人员要面对高度降解的生物学检材。为了解决高度降解DNA检材的个人识别难题,许多实验室采用缩短靶DNA片段的扩增子长度这一基本策略,其中最具代表性的就是miniSTR技术。近年来,一种基于连接酶检测反应(ligase detection reaction,LDR)的单核苷酸多态性(single nucleotide polymorphisms,SNPs)分型技术迅速发展,为解决降解DNA法医学分析难题提供了新的方法。本研究拟构建一套含有8个SNPs位点的基于LDR荧光标记的PCR复合扩增体系,并应用于对甲醛固定石蜡包埋组织(formalin fixed and paraffinembeddedtissues,FFPET)中降解DNA的分型检测。
1 材料与方法
1.1 研究对象
收集6例中国北方汉族的无关个体抗凝血样品(中国医科大学法医物证教研室提供)。选取同一具尸体的心脏、肝脏、脑、肾脏和肺脏组织样品,体积约为2 cm×2 cm×2 cm,10%中性甲醛溶液固定3 d、7 d和15 d后,进行石蜡包埋;抽取心腔血作为阳性对照,以上样品由中国医科大学法医病理学教研室提供。
1.2 试剂和仪器
1.2.1 试剂:Ampligase Thermostable DNA连接酶(美国Epicentre Technologies公司);GeneScan 120 LIZ分子量内标、GeneScan 600 LIZ分子量内标、AmpFℓSTR Identifiler试剂盒和Hi-Di甲酰胺(美国Applied Biosystems公司)。
1.2.2 仪器:PCR扩增仪(美国Applied Biosystems公司,AB9700);遗传分析仪(美国Applied Biosystems公司,AB3500);高速离心机(科大创新股份有限公司中佳分公司,HC-2062);恒温水浴槽(美国Crystal Technology Industries公司,SY-1220);超微量紫外分光光度计(美国Thermo Scientific公司,Nano-Drop2000);超纯水系统(美国Pall Corporation,Cascada LS)。
1.3 DNA提取及DNA质量控制
采用有机酚/氯仿法[1]提取全血检材DNA。FFPET检材经二甲苯脱蜡后[2-3],采用Chelex-100法提取DNA[4]。使用超微量紫外分光光度计测量DNA的OD260和OD280。FFPET检材DNA采用1%琼脂糖凝胶电泳,检测其降解程度,并设计了3对降解程度评价引物,对FFPET检材DNA进行扩增,用以检测其DNA的降解程度。见表1。

表1 降解程度评价引物Tab.1 Primers for evaluating the degradation degree
PCR扩增其他反应参数为:95℃预变性5 min;95℃变性1 min,复性时间为30 s,72℃延伸,延伸时间为20 s,终末72℃延伸10 min,循环34次。PCR扩增体系为:10×PCR缓冲液(含Mg2+)2 μL,dNTP混合物2 μL,上下游引物(5 μmol/L)各2 μL,rTaq酶1 U,模板0.1 μg,补充ddH2O至20 μL。
1.4 SNPs位点的选择
根据相关文献报道和网络数据库(http://www. ncbi.nlm.nih.gov/SNP,http://genome.ucsc.edu/cgi-bin/ hgGateway)中SNPs信息,初步筛选出20个SNPs位点,通过Primer Premier 5.0软件和序列比对工具(http://blast.ncbi.nlm.nih.gov/Blast.cgi)对所有SNPs位点上下游30个核苷酸序列进行自身和相互间结构检查,并进行各SNPs单位点的连接酶检测反应条件的控制与优化,最终确定LDR-PCR复合扩增体系中 的 8个 SNPs位点:rs10802248、rs10516197、rs10488372、 rs2278945、 rs4757318、rs4887255、 rs4889002和rs9304473。
1.5 DNA样品的8个SNPs位点基因型确认
将中国北方地区汉族的个体DNA样品送至宝生物工程(大连)有限公司进行测序,获得8个SNPs位点基因型。
1.6 荧光标记LDR-PCR复合扩增检测的构建
针对各待测SNPs位点分别设计1条公共探针(Ⅰ)和2条等位基因特异性探针(Ⅱ和Ⅲ)。LDR的反应体系为40 μL,其中含10×缓冲液4 μL,LDR探针混合物6 μL,DNA模板0.1 μg,DNA连接酶2 U;反应参数为95℃预变性2 min;95℃变性40 s,63℃退火并连接10 min,循环30次;最后63℃连接20 min;4℃保存。
将连接产物进行PCR扩增,扩增体系为20 μL,其中含10×PCR缓冲液2 μL,dNTP混合物2 μL,Po(5’-GCAAGATAAGGAATCGTAGACT-3’,5 μmol/L)3 μL,Pa(5’-6-FAM-AGGTAACTCCATAAGGT-GTCTT-3’,5 μmol/L)1.25 μL,Pb(5’-6-JOE-AGTGGAATGCTACCAGTTAGAT-3’,5 μmol/L)0.85 μL,Pc(5’-ROX-TCGTGTGATCGTATTGGTTCT-3’,5 μmol/L)0.9 μL,LDR连接产物5 μL,Taq酶1 U;反应参数为:95℃预变性5 min;95℃变性1 min,64℃复性30 s,72℃延伸20 s,循环31次;72℃延伸10 min;4℃保存。PCR产物经AB3500遗传分析仪电泳检测并进行结果分析。根据扩增产物长度和荧光颜色,将SNP位点分为A组、B组和C组。见表2。
2 结果
2.1 DNA降解程度检测结果
心腔血液DNA样品的电泳谱带显示未发生明显降解。经10%中性甲醛溶液固定的各脏器FF-PET DNA谱带均呈现涂抹状弥散,说明DNA发生了降解。固定时间越长,DNA降解程度越高;不同脏器FFPET的DNA降解速率也存在较大差异,见图1。

表2 SNPs位点LDR寡核苷酸探针及扩增产物长度Tab.2 LDR oligonucleotide probes of SNPs and the lengths of PCR products
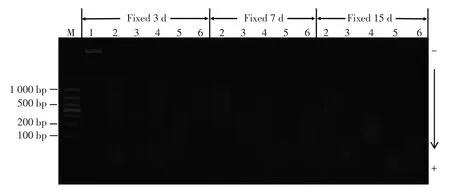
图1 不同固定时间FFPET检材DNA琼脂糖凝胶电泳图谱Fig.1 DNA agarose gel electrophoresis of FFPET fixed for different periods
使用3对扩增产物长度分别为50 bp、103 bp和 198 bp的引物对固定15 d的FFPET检材DNA进行PCR扩增,显示脑组织和心肌组织3种片段长度均能扩出;肺脏和肾脏组织200 bp片段无法扩出;肝脏组织只能扩出50 bp片段,见图2。
2.2 FFPET检材试剂盒直接检测结果

图2 固定15 d的FFPET检材DNA的降解评价引物PCR扩增产物聚丙烯酰氨凝胶电泳图谱Fig.2 PAGE of PCR products obtained from 15⁃day fixed FFPET DNA by using the evaluated primers
应用国际通用的Identifiler试剂盒,对心腔血液和经10%中性甲醛固定15 d的各脏器FFPET检材的DNA样品进行复合扩增。扩增产物经毛细管电泳检测分型。
血液样品所有位点均能成功分型(图3A);肺脏组织样品4个位点检出结果(图3B);肾脏组织样品3个位点检出结果(图3C);肝脏组织样品只有性别位点检出结果(图3D)。
2.3 北方汉族无关个体LDR-PCR复合扩增检测结果
应用荧光标记LDR-PCR复合扩增方法,对6份中国北方地区汉族个体样品进行检测(图4、5),分型结果与测序结果完全一致。
2.4 FFPET样品LDR-PCR复合扩增检测结果
应用LDR-PCR复合扩增体系对10%中性甲醛固定15 d的FFPET检材进行检测,均获得了准确分型结果,见图6。

图3 血液及甲醛固定15 d的FFPET检材经Identifiler试剂盒扩增产物分型图谱Fig.3 Profiles of blood DNA and 15⁃day fixed FFPET DNA using the Identifiler Kit

图4 LDR⁃PCR等位基因分型标准物电泳图谱Fig.4 Profile of the LDR⁃PCR ladder

图5 应用LDR⁃PCR复合扩增方法检测6份中国北方地区汉族无关个体电泳图谱Fig.5 LDR⁃PCR profiles of six Han individuals in Northern China

图6 固定15 d的肺、肾及肝组织FFPET检材LDR⁃PCR复合扩增检测分型图谱Fig.6 LDR⁃PCR profiles of 15⁃day fixed lung,kidney,and liver FFPET DNA
3 讨论
短串联重复序列(short tandem repeats,STR)作为第2代遗传标记,因其基因片段短、扩增效率高、判型准确等特点,现已被广泛地应用在法医学的个人识别和亲缘关系鉴定中。但是在检测降解检材时,常出现等位基因丢失和扩增不均衡等现象。根据降解DNA的特点,可以通过改变引物的结合位置、缩短扩增片段长度等方法来提高基因座的检出率。Applied Biosystems公司推出了miniSTR试剂盒——MiniFiler,其中包含了8个STR基因座和1个性别基因座,扩增产物长度分布在70~283 bp。
SNPs作为第3代遗传标记,被认为是分析降解检材DNA的理想标记,主要是因为其多态性存在于单个核苷酸的碱基类型上。因此与STR相比,能够大大缩短PCR扩增产物的长度。英国法庭科学服务部根据等位基因特异性PCR方法研制出SNPs复合扩增检测试剂盒——Foren-SNPs,用于降解DNA检材的分析[5-6],其中包含20个SNPs位点和1个性别基因座,扩增产物长度分布在57~146 bp。SANCHEZ等[7]筛选出52个SNPs位点用于法医学应用,扩增产物长度在59~115 bp。国际法医遗传学会及欧洲DNA分型学会建立了含有29个SNPs位点的复合扩增系统[8]。Applied Biosystems也推出了SNPs分型试剂盒——GenPlex HID System[9-10],其中包含48个位于常染色体的SNPs位点和1个性别检测位点。FREIRE-ARADAS等[11]选择了18个SNPs位点进行复合扩增,扩增产物长度位于56~118 bp。
为了提高SNPs检测反应的特异性和灵敏度,需要对DNA进行PCR预扩增,大部分扩增产物的长度>70 bp。如果应用LDR技术则不需要进行PCR预扩增,根据紧邻SNPs位点上下游一段核苷酸设计序列特异的寡核苷酸探针,将模板DNA片段长度控制在50 bp以下。ZHANG等[12]设计了21~37 bp长短不等的序列特异性靶序列结合探针,对经DNaseⅠ处理的DNA样品(<100 bp)进行了检测,成功获得了4个SNPs位点的准确分型。
本研究筛选出了8个SNPs位点,成功构建了LDR-PCR复合扩增检测体系,对<50 bp的DNA模板检测获得了准确的分型结果。
综上所述,荧光标记LDR-PCR复合扩增方法适用于高度降解检材的检测,对于<50 bp的DNA片段也可以获得准确的SNPs分型,进而进行法医学个人识别,具有很高的应用价值。
[1]PONCZ M,SOLOWIEJCZYK D,HARPEL B,et al.Construction of human gene libraries from small amounts of peripheral blood:analysis of beta-like globin genes[J].Hemoglobin,1982,6(1):27-36.
[2]SANTOS MC,SAITO CP,LINE SR.Extraction of genomic DNA from paraffin-embedded tissue sections of human fetuses fixed and stored in formalin for long periods[J].Pathol Res Pract,2008,204(9):633-636.DOI:10.1016/j.prp.2008.04.005.
[3]吕晋,王慧君,刘超,等.福尔马林固定石蜡包埋组织3种DNA提取方法比较[J].中国法医学杂志,2013,28(3):239-241.DOI:10.13618/J.ISSN.1001-5728.2013.03.019.
[4]WALSH PS,METZGER DA,HIGUCHI R.Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material[J].Biotechniques,1991,10(4):506-513.
[5]DIXON LA,MURRAY CM,ARCHER EJ,et al.Validation of a 21-locus autosomal SNP multiplex for forensic identification purposes[J].Forensic Sci Int,2005,154(1):62-77.DOI:10.1016/j.forsciint. 2004.12.011.
[6]DIXON LA,DOBBINS AE,PULKER HK,et al.Analysis of artificially degraded DNA using STRs and SNPs-results of a collaborative European(EDNAP)exercise[J].Forensic Sci Int,2006,164(1):33-44.DOI:10.1016/j.forsciint.2005.11.011.
[7]SANCHEZ JJ,PHILLIPS B,BØRSTING C,et al.A multiplex assay with 52 single nucleotide polymorphisms for human identification[J].Electrophoresis,2006,27(9):1713-1724.DOI:10.1002/elps. 200500671.
[8]SANCHEZ JJ,BØRSTING C,BALOGH K,et al.Forensic typing of autosomal SNPs with a 29 SNP-multiplex-results of a collaborative EDNAP exercise[J].Forensic Sci Int Genet,2008,2(3):176-183. DOI:10.1016/j.fsigen.2007.12.002.
[9]TOMAS C,STANGEGAARD M,BØRSTING C,et al.Typing of 48 autosomal SNPs and amelogenin with GenPlex SNP genotyping system in forensic genetics[J].Forensic Sci Int Genet,2008,3(1):1-6.DOI:10.1016/j.fsigen.2008.06.007.
[10]TOMAS C,BØRSTING C,MORLING N,et al.A 48-plex autosomal SNP GenPlex™assay for human individualization and relationship testing[J].Methods Mol Biol,2012,830:73-85.DOI:10. 1007/978-1-61779-461-2_6.
[11]FREIRE-ARADAS A,FONDEVILA M,KRIEGEL A K,et al.A new SNP assay for identi fi cation of highly degraded human DNA[J].Forensic Sci Int Genet,2012,6(3):341-349.DOI:10.1016/j. fsigen.2011.07.010.
[12]ZHANG Z,WANG BJ,GUAN HY,et al.A LDR-PCR approach for multiplex polymorphisms genotyping of severely degraded DNA with fragment sizes<100 bp[J].J Forensic Sci,2009,54(6):1304-1309.DOI:10.1111/j.1556-4029.2009.01166.x.
(编辑 于 溪)
Study on SNP Genotyping of Degraded DNA by Fluorescence⁃labeled Multiplex LDR⁃PCR Amplification
XING Jiaxin1,SUN Yihua1,2,XUAN Jinfeng1,YAO Jun1,DING Mei1,PANG Hao1,LI Chunmei1,XIA Xi1,WANG Baojie1
(1.Department of Forensic Genetics,School of Forensic Medicine,China Medical University,Shenyang 110122,China;2.Forensic DNA Section,Wuzhong District Bureau of Suzhou Public Security Bureau,Suzhou 215104,China)
ObjectiveIn this study,a multiplex PCR amplification system was constructed based on fluorescent labeling PCR and LDR,to provide a new strategy for analyzing severely degraded DNA.MethodsEight SNP loci(rs10802248,rs10516197,rs10488372,rs2278945,rs4757318,rs4887255,rs4889002,and rs9304473)were selected.Their LDR probes and PCR primers of linked products were designed and synthesized.Ligase detection reaction,PCR amplification,and capillary gel electrophoresis(CEG)were performed to establish the multiplex LDRPCR amplification system.ResultsThe genotypes of these 8 loci were obtained simultaneously by the fluorescence-labeled multiplex LDR-PCR amplification method.The loci profiles obtained by fluorescence-labeled multiplex LDR-PCR amplification were in accordance with those obtained by direct sequencing of the polymorphic regions in samples from all individuals.By fluorescence-labeled multiplex LDR-PCR amplification,the 8 SNP loci were efficiently amplified from the severely degraded FFPET DNA.ConclusionEight SNP loci results could be obtained simultaneously by using the multiplex LDR-PCR amplification system,which is a simple,efficient,and practical SNP genotyping method with accurate and reliable results for highly degraded samples.
forensics biological evidence;ligase detection reaction;compound amplification;degraded DNA;SNPs
R89
A
0258-4646(2017)08-0703-07
10.12007/j.issn.0258-4646.2017.08.008
辽宁省教育厅科学研究一般项目(L2013319)
邢佳鑫(1980-),男,讲师,博士.
王保捷,E-mail:bjwang@cmu.edu.cn
2016-12-22
网络出版时间:
猜你喜欢
探索科学(学术版)(2021年4期)2021-05-20
小哥白尼(野生动物)(2021年10期)2021-02-12
法制博览(2019年32期)2019-11-22
中国中医急症(2019年10期)2019-05-21
求学·文科版(2018年5期)2018-07-12
中华骨与关节外科杂志(2017年1期)2017-05-17
法医学杂志(2016年3期)2016-07-22
课堂内外(初中版)(2015年2期)2015-09-10
智能制造(2015年4期)2015-05-12
法制与社会(2009年26期)2010-06-29
