
以p73为靶点的抗肿瘤药物研究进展
2017-08-29 11:32牟汉川杨志宽暴亚锋张继虹
中国药理学通报 2017年9期
牟汉川,杨志宽,暴亚锋,张继虹
(昆明理工大学医学院衰老与肿瘤分子遗传学实验室,云南 昆明 650500)
以p73为靶点的抗肿瘤药物研究进展
牟汉川,杨志宽,暴亚锋,张继虹
(昆明理工大学医学院衰老与肿瘤分子遗传学实验室,云南 昆明 650500)
肿瘤抑制因子p73可转录形成具有促凋亡(TAp73)和抗凋亡(△Np73)功能的2种亚型。但是,在肿瘤进程中,TAp73-负调节因子(如△Np73、突变p53、MDM2和iASPP)复合体的形成会阻碍TAp73的肿瘤抑制活性。因此,通过靶向抑制负调节因子或破坏TAp73-负调节因子复合体,释放出TAp73,进而达到抑制肿瘤的目的。该文就p73的靶向调控及相关药物的研究进展进行综述。
肿瘤抑制因子;p73;TAp73;凋亡;负调节因子;复合体
p73是一个转录因子,属于p53蛋白家族。肿瘤抑制因子p73可参与细胞增殖、分化、发育和凋亡的调控。同时,p73在神经发育和分化[1]、代谢调控[2]、精子发生和雄性生育能力调控[3]中发挥其独特的功能。通过选择性使用启动子P1或P2,p73可形成全长型TAp73(full-length transactivation,TA)和N端截短型△Np73(N-terminally truncated,△N)2种亚型。TAp73作为肿瘤抑制因子可诱导细胞凋亡和周期阻滞。相反,△Np73具有癌基因功能,可抑制野生型p53和TAp73的功能。在肿瘤进程中,p73几乎不受到突变的影响,同时具有与p53类似的抗肿瘤活性[4]。因而在p53突变或失活的肿瘤中,p73可替代p53发挥其肿瘤抑制因子的作用。因此,可通过操控p53家族成员p73来治疗肿瘤。本文将着重阐述以p73为靶点的药物研究进展,并讨论相关化合物在肿瘤治疗中的作用机制。
1 p73与肿瘤的关系
p53蛋白家族在结构和功能上具有相似性,例如,在作为基因转录正向或负向调节因子结合位点的N端转录激活区(transcription activation domain,TAD)、与靶基因应答元件相结合的DNA结合区(DNA-binding domain,DBD)、频繁发生基因剪接和翻译后修饰的C端低聚体区(oligomerization domain,OD)都具有很高的序列同源性。同时,p73可激活p53靶基因的表达,如p21、PUMA、NOXA、IGF-BP3和Cyclin G,从而发挥与p53相似的肿瘤抑制功能[5]。
p53基因家族含有2个启动子,P1启动子位于序列上游1号外显子,P2启动子位于3号外显子(p63和p73)或4号外显子(p53)。通过选择性使用启动子和选择性切割可编码形成不同的亚型(Fig 1)。从P1启动转录形成包含转录激活区的全长亚型TA型(TAp63和TAp73),它们都具有与野生型p53相似的肿瘤抑制功能。很多DNA损伤药物都可诱导TAp73的产生,并且使用能与目的蛋白形成复合体并抑制其功能的负显性调节蛋白或干扰RNA来阻滞TAp73的功能而增强肿瘤细胞的化疗耐药。从P2启动转录形成不包含转录激活区的截短亚型△N型(△Np53、△Np63和△Np73),具有癌基因功能,可负显性调节野生型p53、TAp63和TAp73,并抑制其靶基因的激活,促进肿瘤进程[6]。同时,△Np63和△Np73在表皮发育(△Np63)或神经发育(△Np73)中发挥重要作用[7]。选择性切割大多数发生在序列3′端的10~13号外显子,形成α、β和γ等亚型。

Fig 1 Structure of p53 family members
肿瘤抑制因子p73主要存在功能相反的2种亚型TAp73和△Np73。在小鼠模型中整体敲除p73,只表现出发育异常,比如生殖障碍、交感神经元丢失和慢性感染和炎症,没有肿瘤的形成[8]。但是,在小鼠中特异敲除TAp73,发现TAp73-/-小鼠肿瘤易感性增加,并且在致癌物的诱导下,70%的敲除小鼠形成肿瘤[9]。相反,△Np73-/-小鼠可抑制肿瘤的生长,诱导成瘤的几率很低[10]。研究发现,在乳腺癌、卵巢癌、前列腺癌、肺癌、结肠癌、皮肤癌和肝癌等肿瘤病例中,△Np73都有高表达,同时,在大多数肿瘤中还伴随着TAp73的高表达[11]。Slade等[12]发现TAp73可转录激活△Np73的表达,然后,△Np73竞争性结合TAp73的C端低聚体区,抑制TAp73的转录激活功能,使其丢失肿瘤抑制功能。
在大多数实体瘤中,除了△Np73可抑制TAp73的抗肿瘤活性外,还有其他负调节因子参与到TAp73的负显性调控中(Fig 2)。这些负调节因子主要可分为两类,一类为p53家族蛋白,如△Np63、△Np73和突变p53。Rocco等[13]发现,在头颈鳞状细胞癌中,△Np63作为肿瘤存活因子,它通过与TAp73的PUMA启动子区结合,抑制TAp73依赖的凋亡,从而促进肿瘤的生存。Strano等[14]通过体外和体内实验证实,突变p53一方面与TAp73的C端低聚体区结合,干扰TAp73同质多聚体的形成,另一方面与TAp73的DNA结合区结合,妨碍TAp73结合DNA,从而导致TAp73不能转录激活下游靶基因的表达。另一类为非p53家族相关蛋白,例如MDM2、iASPP和mTOR。Zeng等[15]发现在p53缺失的细胞中,过表达p73可上调MDM2的表达,然后MDM2通过与p300竞争性结合p73的N端区域而抑制p73的转录活性。但是,并不会诱导p73通过MDM2介导的泛素化途径降解。因此,在肿瘤治疗中,可直接靶向抑制这些负调节因子,或破坏TAp73与其负调节因子的相互作用,从而打破负调节因子与TAp73之间的平衡,释放出TAp73,以重新激活TAp73的抗肿瘤活性。
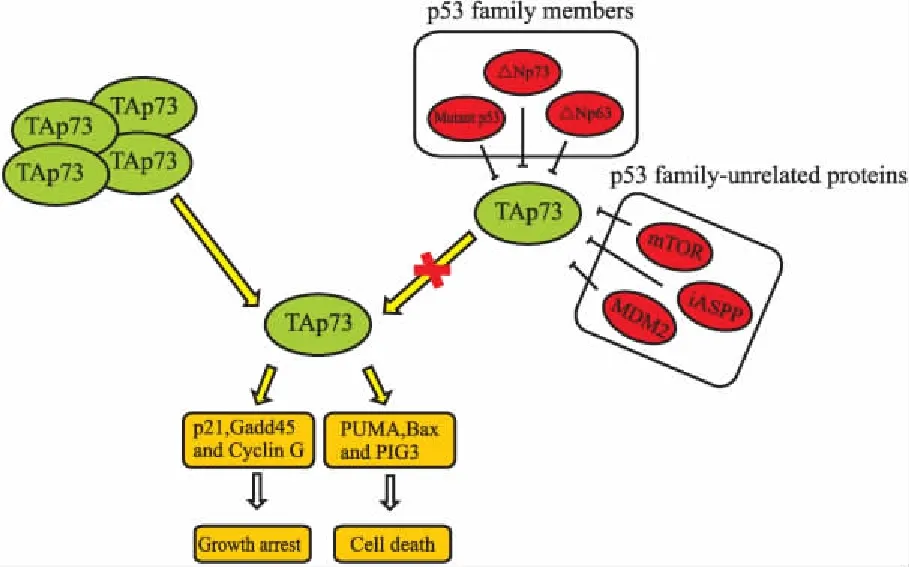
Fig 2 Inhibition and activation of TAp73 in tumor cells
2 靶向作用TAp73的药物研究
研究表明,很多药物可诱导肿瘤抑制因子TAp73的产生。目前,激活TAp73的策略主要有以下3种:① 增加TAp73的表达,如激活TAp73的转录或抑制TAp73的降解;② 调节TAp73上游调节因子的表达,比如上调激酶和乙酰转移酶的活性;③ 破坏TAp73与其负调节因子的相互作用。
Nutlin-3是MDM2的小分子抑制剂,可阻断MDM2-p53的相互作用,稳定p53,诱导p53依赖的细胞周期阻滞和凋亡。在野生型p53的肿瘤中,Nutlin-3通过稳定p53发挥其抗肿瘤作用。但是,在p53缺失或突变的肿瘤细胞中,由于p73具有与p53高度同源的N端MDM2结合区,因此,Nutlin-3可通过抑制MDM2-p73复合体的形成,增强p73的转录活性和稳定性,一方面启动TAp73的转录,使其在p53缺失的肿瘤细胞中积累,促进凋亡相关的蛋白NOXA、PUMA和Siva-1的表达,诱导细胞凋亡[16];另一方面,增强E2F-1、MDMX和IKK-α竞争性结合TAp73的N端MDM2结合区,稳定p73并延长TAp73的半衰期[17]。
全部△Np73的亚型通过与TAp73形成异聚复合体或与TAp73竞争C端低聚体区,进而负显性调节TAp73依赖的生长抑制和凋亡效应。在早期研究中,环氧化酶抑制剂Celecoxib作为抗炎药发挥其功能。Lau等[18]研究发现,在神经母细胞瘤中,Celecoxib通过干扰转录因子E2F-1与p73启动子的结合,下调△Np73的表达,上调TAp73β的表达,改变TA/△Np73的比例,诱导p57、PUMA和NOXA的产生,进而导致TAp73依赖的凋亡。另外,在肝癌患者中,通过下调EGFR配体双调蛋白的自分泌或旁分泌环路,可明显降低△Np73的积累,同时再激活肝癌病人中TAp73的表达,进而改变TA/△Np73的比例可以在临床中取得良好的治疗效果[19]。Venkatanarayan等[20]通过调控p53家族成员p63和p73治疗p53突变的肿瘤取得新的进展,研究显示,普兰林肽(pramlintide)可特异抑制p53缺陷小鼠中△Np73和△Np63的表达,随之上调TAp63和TAp73的表达,加速肿瘤的退化并增加小鼠的生存期。同时,上调代谢相关调节因子IAPP、GLS2和TIGAR启动凋亡。
突变p53结合TAp73并使其失活,已经认为是突变p53获得性功能的一部分[21]。因此,靶向破坏突变p53-TAp73复合体可用于治疗肿瘤。Zhang等[22]通过高通量筛选获得既能降解突变p53,又能通过激活TAp73的表达而活化野生型p53信号通路的小分子化合物NSC59984。研究发现,在突变p53背景下,NSC59984破坏TAp73-突变p53复合体后,一方面触发MDM2介导的泛素化途径,降解突变p53,另一方面释放出TAp73,上调野生型p53信号通路中p21、DR5、PUMA和NOXA的表达,诱导TAp73依赖的肿瘤细胞死亡。同时,抑制小鼠移植瘤的生长。NSC59984在细胞水平和动物模型中都取得不错的进展。Di Agostino等[23]以突变p53-TAp73异二聚体DNA结合区的结构模型为基础,设计出干扰短肽(short interfering mutant p53 peptides,SIMPs)与TAp73竞争性结合突变p53,促使TAp73累积并启动凋亡级联反应。
p53凋亡刺激蛋白(ASPP)家族由ASPP1、ASPP2和iASPP 3个成员组成。其中,ASPP1和ASPP2能结合p53、p63和p73的核心区,启动凋亡相关基因BAX、PIG-3和IGF-BP3的转录,但是iASPP作为p53家族的负调节因子发挥其功能。根据p53的118~142残基和171~181残基合成出的凋亡肽37AA,在p53失活的肿瘤中,通过特异结合iASPP而干扰TAp73和iASPP的相互作用,使胞内TAp73去阻遏并恢复其功能,进而导致TAp73调节的PUMA、p21和DR5基因激活[24]。
mTOR是一个丝氨酸/苏氨酸激酶,在细胞生长和增殖中发挥重要作用。Rosenbluth等[25]研究发现mTOR还参与p73的调节,通过使用TAp73基因标签进行高通量筛选得到能激活TAp73表达的候选化合物。分析后发现,在乳腺癌细胞中,使用mTOR抑制剂,比如雷帕霉素(rapamycin)和二甲双胍(metformin),可激活TAp73,上调胰岛素受体基因(insulin receptor,INSR)、黄嘌呤脱氢酶基因(xanthine dehydrogenase,XDH)、结节性硬化症基因(tuberous sclerosis 1,TSC1)的表达,诱导细胞发生凋亡。但是,在乳腺癌细胞中敲低p73后使用mTOR抑制剂,阻止了细胞凋亡的发生。这表明在细胞增殖中mTOR与p73存在调节关系。因此,在p53失活的肿瘤中,能通过抑制mTOR激活TAp73而抑制肿瘤。并且,在临床应用中,mTOR抑制剂上调TAp73的表达后,可增加顺铂对乳腺癌治疗的敏感性[26]。
3 其他靶向策略
除了破坏TAp73-负调节因子复合体激活TAp73外,还可通过凋亡切割和microRNAs等其他方式调控TAp73的功能。最新研究显示[27],多西紫杉醇(docetaxel,DOC)通过caspase-3切割TAp73α,形成切割片段并进入细胞核内,诱导乳腺癌细胞失巢凋亡(anoikis)的发生。Sayan等[28]发现,DNA损伤应激后TAp73被caspase-3和caspase-8切割,切割片段进入线粒体启动肿瘤坏死因子相关凋亡配体(tumor necrosis factor-related apoptosis-inducing ligand,TRAIL)诱导的凋亡反应。研究表明,miRNA与p73依赖的化疗敏感性相关。Sampath等[29]通过去乙酰化酶抑制剂诱导miR-106b的上调,降低E3泛素连接酶Itch的表达,导致其凋亡底物TAp73的积累,进而启动慢性淋巴白血病(chronic lymphocytic leukemia,CLL)中的凋亡相关基因PUMA的表达。Alla等[30]研究发现,在转移性肿瘤中通常高表达E2F1,导致△Np73的表达上调,进而抑制miR-205的表达。在肿瘤中,活化E2F1/△Np73/miR-205反馈回路可直接导致化疗耐药、不良预后和促进肿瘤进程。因此,可通过药物或其他治疗手段诱导miR-205的高表达来负向调节E2F1/△Np73/miR-205信号通路,可达到治疗晚期肿瘤的目的。
此外,在急性淋巴细胞白血病(acute lymphoblastic leukemia,ALL)中,TAp73启动子区的CpG岛发生超甲基化能沉默TAp73的表达,同时,也与其较差的临床预后相关[31]。Liang等[32]发现DNA甲基转移酶抑制剂5-aza-2′-deoxycytidine(5-Aza-CdR),能促进TAp73启动子去甲基化,激活TAp73的转录和蛋白的表达水平,进而抑制肿瘤的发生发展。同时,在乳腺癌中,TAp73启动子的去甲基化可增强对化疗的敏感性[33]。
4 总结与展望
深入研究发现人类肿瘤中存在复杂的突变p53背景,靶向突变p53的治疗策略将面临巨大挑战[34]。因此,探究新的有效的治疗p53突变肿瘤的方法已十分迫切[35]。通过调控p53家族成员p73治疗p53缺失或突变的肿瘤为临床应用研究提供新的方向。在p53失活的情况下,通过激活p73来替代p53发挥其抗肿瘤功能。值得注意的是,在药物或其他治疗手段诱导肿瘤抑制因子TAp73的表达时,能启动p53非依赖性的凋亡级联效应,进而抑制肿瘤进程。但是,靶向p73化合物及其作用机制还有待进一步的研究。例如,使用其他药物与p73靶向药物联合进行治疗,增强药物对p53缺失或突变肿瘤的疗效,同时,降低对正常细胞和组织的毒副作用。另外,靶向药物的特异性还有待提高。目前,以p73为靶点的药物研究将成为肿瘤治疗新的焦点,发现靶向p73的药物将充满机遇与挑战。
[1] Killick R, Niklison-Chirou M, Tomasini R, et al. p73: a multifunctional protein in neurobiology[J].MolNeurobiol, 2011,43(2): 139-46.
[2] Cutruzzola F, Avigliano L, Candi E. p73 keeps metabolic control in balance[J].CellCycle, 2014,13(2): 179-80.
[3] Inoue S, Tomasini R, Rufini A, et al. TAp73 is required for spermatogenesis and the maintenance of male fertility[J].ProcNatlAcadSciUSA, 2014,111(5): 1843-8.
[4] Ferraiuolo M, Di Agostino S, Blandino G, et al. Oncogenic intra-p53 family member interactions in human cancers[J].FrontOncol, 2016,6: 77.
[5] Zhu J, Jiang J, Zhou W, et al. The potential tumor suppressor p73 differentially regulates cellular p53 target genes[J].CancerRes,1998,58(22): 5061-5.
[6] Inoue K, Fry E A. Alterations of p63 and p73 in human cancers[J].SubcellBiochem, 2014,85: 17-40.
[7] Dötsch V, Bernassola F, Coutandin D, et al. p63 and p73, the ancestors of p53[J].ColdSpringHarbPerspectBiol, 2010,2(9): a004887.
[8] Yang A, Walker N, Bronson R, et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours[J].Nature, 2000,404(6773): 99-103.
[9] Tomasini R, Tsuchihara K, Wilhelm M, et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions[J].GenesDev, 2008,22(19): 2677-91.
[10]Wilhelm M T, Rufini A, Wetzel M K, et al. Isoform-specific p73 knockout mice reveal a novel role for delta Np73 in the DNA damage response pathway[J].GenesDev, 2010,24(6): 549-60.
[11]Engelmann D, Meier C, Alla V, et al. A balancing act: orchestrating amino-truncated and full-length p73 variants as decisive factors in cancer progression[J].Oncogene, 2015,34(33): 4287-99.
[12]Slade N, Zaika A I, Erster S, et al. DeltaNp73 stabilises TAp73 proteins but compromises their function due to inhibitory hetero-oligomer formation[J].CellDeathDiffer, 2004,11(3): 357-60.
[13]Rocco J W, Leong C O, Kuperwasser N, et al. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis[J].CancerCell, 2006,9(1): 45-56.
[14]Strano S, Munarriz E, Rossi M, et al. Physical and functional interaction between p53 mutants and different isoforms of p73[J].JBiolChem, 2000,275(38): 29503-12.
[15]Zeng X, Chen L, Jost C A, et al. MDM2 suppresses p73 function without promoting p73 degradation[J].MolCellBiol,1999,19(5): 3257-66.
[16]Ray R M, Bhattacharya S, Johnson L R. Mdm2 inhibition induces apoptosis in p53 deficient human colon cancer cells by activating p73-and E2F1-mediated expression of PUMA and Siva-1[J].Apoptosis, 2011,16(1): 35-44.
[17]Lau L M, Nugent J K, Zhao X, et al. HDM2 antagonist Nutlin-3 disrupts p73-HDM2 binding and enhances p73 function[J].Oncogene, 2008,27(7): 997-1003.
[18]Lau L M, Wolter J K, Lau J T, et al. Cyclooxygenase inhibitors differentially modulate p73 isoforms in neuroblastoma[J].Oncogene, 2009,28(19): 2024-33.
[19]Castillo J, Goni S, Latasa M U, et al. Amphiregulin induces the alternative splicing of p73 into its oncogenic isoform DeltaEx2p73 in human hepatocellular tumors[J].Gastroenterology, 2009,137(5): 1805-15.
[20]Venkatanarayan A, Raulji P, Norton W, et al. Novel therapeutic interventions for p53-altered tumors through manipulation of its family members, p63 and p73[J].CellCycle, 2016,15(2): 164-71.
[21]Stindt M H, Muller P A, Ludwig R L, et al. Functional interplay between MDM2, p63/p73 and mutant p53[J].Oncogene, 2015,34(33): 4300-10.
[22]Zhang S, Zhou L, Hong B, et al. Small-molecule NSC59984 restores p53 pathway signaling and antitumor effects against colorectal cancer via p73 activation and degradation of mutant p53[J].CancerRes, 2015,75(18): 3842-52.
[23]Di Agostino S, Cortese G, Monti O, et al. The disruption of the protein complex mutantp53/p73 increases selectively the response of tumor cells to anticancer drugs[J].CellCycle, 2008,7(21): 3440-7.
[24]Bell H S, Dufes C, O′Prey J, et al. A p53-derived apoptotic peptide derepresses p73 to cause tumor regressioninvivo[J].JClinInvest, 2007,117(4): 1008-18.
[25]Rosenbluth J M, Mays D J, Pino M F, et al. A gene signature-based approach identifies mTOR as a regulator of p73[J].MolCellBiol, 2008,28(19): 5951-64.
[26]Wong S W, Tiong K H, Kong W Y, et al. Rapamycin synergizes cisplatin sensitivity in basal-like breast cancer cells through up-regulation of p73[J].BreastCancerResTreat, 2011,128(2): 301-13.
[27]Alsafadi S, Tourpin S, Bessoltane N, et al. Nuclear localization of the caspase-3-cleaved form of p73 in anoikis[J].Oncotarget, 2016,7(11): 12331-43.
[28]Sayan A E, Sayan B S, Gogvadze V, et al. P73 and caspase-cleaved p73 fragments localize to mitochondria and augment TRAIL-induced apoptosis[J].Oncogene, 2008,27(31): 4363-72.
[29]Sampath D, Calin G A, Puduvalli V K, et al. Specific activation of microRNA106b enables the p73 apoptotic response in chronic lymphocytic leukemia by targeting the ubiquitin ligase Itch for degradation[J].Blood, 2009,113(16): 3744-53.
[30]Alla V, Kowtharapu B S, Engelmann D, et al. E2F1 confers anticancer drug resistance by targeting ABC transporter family members and Bcl-2 via the p73/DNp73-miR-205 circuitry[J].CellCycle, 2012,11(16): 3067-78.
[31]Roman-Gomez J, Jimenez-Velasco A, Castillejo J A, et al. Promoter hypermethylation of cancer-related genes: a strong independent prognostic factor in acute lymphoblastic leukemia[J].Blood, 2004,104(8): 2492-8.
[32]Liang W, Xia H, Li J, et al. 5-Aza-2′-deoxycytidine increases the sensitivity of human bone marrow mesenchymal stem cells to chemotherapeutic agents by demethylation of p73[J].JPediatrHematolOncol, 2012,34(2): 108-15.
[33]Lai J, Yang F, Zhang W, et al. TAp73 and deltaNp73 have opposing roles in 5-aza-2′-deoxycytidine-induced apoptosis in breast cancer cells[J].MolCells, 2014,37(8): 605-12.
[34]Aschauer L, Muller P A. Novel targets and interaction partners of mutant p53 gain-of-function[J].BiochemSocTrans, 2016,44(2): 460-6.
[35]代晓丽, 刘 静, 罗 瑛, 张继虹. 靶向作用p53 药物的研究进展[J].中国药理学通报, 2014,30(7): 912-6.
[35]Dai X L, Liu J, Luo Y, Zhang J H. Research progress on drugs targeting p53[J].ChinPharmacolBull, 2014,30(7): 912-6.
Advancesinp73-targetinganti-tumordrugs
MOU Han-chuan, YANG Zhi-kuan, BAO Ya-feng, ZHANG Ji-hong
(LabofMolecularGeneticsofAgingandTumor,FacultyofMedicine,KunmingUniversityofScienceandTechnology,Kunming650500,China)
The transcription factor p73 belongs to the p53 family of tumor suppressors, and can be transcribed into different isoforms with either pro- or anti-apoptotic(TAp73 and △Np73) functions. However, the tumor suppressor activity of TAp73 is inhibited through complex formation with inhibitory proteins(e.g. △Np73, mutant p53, MDM2 and iASPP). Therefore, it is a kind of tumor therapy strategy to reactivate TAp73 through targeting these inhibitors directly or release TAp73 from the complex by targeting their interaction. This review discusses the possible strategies of targeting p73 for its reactivation and the acting mechanism of related compounds.
tumor suppressor; p73; TAp73; apoptosis; inhibitory proteins; complex
时间:2017-8-20 16:47 网络出版地址:http://kns.cnki.net/kcms/detail/34.1086.R.20170820.1647.012.html
2017-04-20,
2017-05-15
国家自然科学基金资助项目(No 81560601);昆明理工大学分析测试基金项目(No 2016M2014236022)
牟汉川(1992-),男,硕士生,研究方向:肿瘤药理学,E-mail:norton2016@126.com; 张继虹(1972-),女,博士,教授,硕士生导师,研究方向:分子药理学及抗肿瘤药物机制,通讯作者,E-mail:zhjihong2000@126.com
10.3969/j.issn.1001-1978.2017.09.006
A
:1001-1978(2017)09-1207-04
R-05;R341;R977.6;R979.1
猜你喜欢
农业工程学报(2022年13期)2022-10-09
中老年保健(2022年1期)2022-08-17
保健医苑(2022年5期)2022-06-10
保健医苑(2022年4期)2022-05-05
作物学报(2022年3期)2022-01-22
汽车维修与保养(2021年8期)2021-02-16
中华养生保健(2020年5期)2020-11-16
中华建设(2019年7期)2019-08-27
中华肩肘外科电子杂志(2019年4期)2019-08-24
医学研究杂志(2015年7期)2015-06-22
