
Aberrant control of NF-κB in cancer permits transcriptional and phenotypic plasticity, to curtail dependence on host tissue: molecular mode
2017-08-27 03:24:20SpirosVlahopoulos
Cancer Biology & Medicine 2017年3期
Spiros A. Vlahopoulos
The First Department of Pediatrics, University of Athens, Horemeio Research Laboratory, Athens 11527, Greece
Aberrant control of NF-κB in cancer permits transcriptional and phenotypic plasticity, to curtail dependence on host tissue: molecular mode
Spiros A. Vlahopoulos
The First Department of Pediatrics, University of Athens, Horemeio Research Laboratory, Athens 11527, Greece
The role of the transcription factor NF-κB in shaping the cancer microenvironment is becoming increasingly clear. Inflammation alters the activity of enzymes that modulate NF-κB function, and causes extensive changes in genomic chromatin that ultimately drastically alter cell-specific gene expression. NF-κB regulates the expression of cytokines and adhesion factors that control interactions among adjacent cells. As such, NF-κB fine tunes tissue cellular composition, as well as tissues' interactions with the immune system. Therefore, NF-κB changes the cell response to hormones and to contact with neighboring cells. Activating NF-κB confers transcriptional and phenotypic plasticity to a cell and thereby enables profound local changes in tissue function and composition. Research suggests that the regulation of NF-κB target genes is specifically altered in cancer. Such alterations occur not only due to mutations of NF-κB regulatory proteins, but also because of changes in the activity of specific proteostatic modules and metabolic pathways. This article describes the molecular mode of NF-κB regulation with a few characteristic examples of target genes.
Cytokine; mucin; chemokine; IL-8/CXCL8; MUC1; NF-κB; IL-6; TNFα
Introduction
NF-κB is an inducible transcription factor that recognizes and binds specific DNA sequences. It enables cells to adapt to tissue homeostatic changes by modulating genomic function1. NF-κB is a dimer of Rel proteins. These Rel proteins are regulated by assorted post-translational modifications, as well as interactions with other proteins, especially inhibitor of κB (IκB). IκB keeps the Rel dimer in a latent state in the cytoplasm1. IκBα is encoded by an NF-κB-regulated gene, reflecting the spectrum and complexity of NF-κB effects on the cell. IκBα may sequester tumor suppressor p53 and operate in the mitochondria by blocking the apoptotic mediator Bax depending on the presence of protein scaffolds2,3. NF-κB is activated by discrete, cellspecific post-translational modifications, and enters the nucleus and mitochondria to alter the expression of multiple gene clusters4-7.
These clusters include different gene types: those that facilitate drastic changes in cell phenotype, promote cell survival, or initiate cell death. Given the cell lineage and genomic state, NF-κB target genes change the cell fate depending on the cell’s specific contribution to tissue structure and function8-10. These changes could be permanent, last over several cell divisions, or be readily reversible depending on the enzymes that accompany transcription factor complexes on chromatin, as well as the ensuing physical or chemical DNA changes. NF-κB target genes include several potent modulators of the immune system that link tissue homeostasis to all the organism functions11-14. Notably, various NF-κB target genes participate in negative feedback mechanisms that limit the key functions of NF-κB and thereby help control inflammation. In cancer, aberrant cellular response to NF-κB activation alters a cell’s phenotype, local microenvironment, and communication with the immune system5.
Cycle of NF-κB activity
Specifically, canonical NF-κB is composed of a Rel protein dimer, which is maintained in its latent cytoplasmic form by IκBα15,16. Cell stress signals and diverse hormonal andmetabolic mediators induce kinase IKKβ to phosphorylate IκBα. This process commonly leads to the proteasomal degradation of IκBα. The freed Rel dimer then enters the nucleus and binds to specific DNA sites that function as cellstress response elements. The Rel dimer also binds to DNA within densely packed heterochromatin17. The enzymes that are associated with the Rel dimer remodel chromatin and change target gene expression depending on the activities of other transcription factors and the type of post-translational modifications on the Rel dimer itself18-20. In particular, NF-κB recruits and redistributes proteins that enhance gene expression over a distance, such as MED1, to interact with chromatin and facilitate promoter-enhancer interactions7,21. A cell typically coordinates the expression of differentiationsteering genes through hundreds of distal enhancers that contain dense binding sites to transcription factors. These enhancers are termed as super-enhancers and are extensively replaced during NF-κB activation; they allow inflammatory gene expression to steer cellular function8.
However, NF-κB also induces the expression of IκBα, which is encoded by a gene that does not require de novo chromatin remodeling. This process generates a negative feedback response that terminates the expression of potent inflammatory mediators22,23. A cell contains several proteolytic systems that can degrade IκBα. In a cancer cell, cytotoxic drugs induce cell stress, which then changes cellular proteostasis, including IκBα proteolysis24. Drastic metabolite changes sufficient to alter enzymatic activity in cellular compartments involved in protein turnover induce NF-κB to facilitate cellular responses to these changes25. Under certain tissue homeostatic conditions, drugs activate NF-κB dimers that facilitate cell survival, express proteins that protect the cancer cell from the immune system, and enable metastasis26. To add complexity, in some drug-resistant cancer cell types, Rel dimers are induced by noncanonical signaling pathways and control the expression of developmental genes not restricted by IκBα. In such cases, the main transducer for noncanonical NF-κB activity is the I-kappa B kinase subunit alpha (IKKα)27. These cells are resistant to drugs that inhibit the upstream inducers of canonical signaling5.
To deliver long-term effects, NF-κB can induce the expression of the methyltransferase DNMT1. DNMT1 can inhibit the expression of tumor suppressor genes over many cell divisions by methylating the CpG islands on the genes28. A tissue regulates inflammation through multiple mechanisms with the following central theme: substances related to infection, injury, or tissue damage by other sources activate the immune system and inhibit tissue function and tissue growth, thereby yielding bioenergetic priority to the immune response7. Inflammatory mediators regulate one another through diverse mechanisms and consequently sustain the immune response in proportion to the stimuli that signal a localized disruption in tissue homeostasis8. Finally, inflammatory mediators gradually disappear and local cells activate signals that re-establish tissue function29(Figure 1).
This chain of events is disrupted in cancer, but key control elements may remain functional, as shown by the effects of signal interference on neoplastic tissue26,30,31. Importantly, one must recognize that feedback signaling to NF-κB in cancer is disrupted to interfere with the fundamental functions of tumor suppressor genes. In brief, cell survival control is not intertwined with tissue homeostasis5. Intracellular turnover controls NF-κB activity, which in turn controls cellular communication with the rest of the organism through cytokines24. Malignant cells increase intracellular turnover, which then enables the NF-κB-driven expression of antiapoptotic factors and the degradation of pro-apoptotic proteins24,32. This occurrence permits cancer cell survival in abnormal locations, but disrupts the tissue feedback with the immune system essential to optimum tissue growth, development, and function5,33.
Molecular types induced by NF-κB
In tissue development, NF-κB interacts with two protein types: inflammatory and tissue regeneration mediators10,34. In oncogenesis, the proteins that regulate inflammation can interact with oncogenes or tumor suppressors. The proteins that mediate tissue regeneration mostly appear in the expression signatures of tumors. However, these proteins may also assume an oncosuppressive role in certain cellular subsets of inflammatory cancers35-37.
Specifically, NF-κB cooperates with transcription factors, such as AP-1, to induce the expression of genes that encode inflammatory cytokines, adhesion molecules, and extracellular proteases (Figure 2)38. These gene products serve essential functions during inflammation and may induce apoptosis in tumors. AP-1 proteins function as dimers that form coiled coils upon dimerization; they belong to the broader class of basic leucine zipper (bZip) transcription factors. These bZip transcription factors include the activating transcription factor (ATF) family. ATF dimers regulate cell growth and facilitate the response to cell cycle checkpoint enzymes by activating cyclin gene expression39. This activity of AP-1 and ATF links the progress of processes, such as DNA repair, with cell cycle progression40.
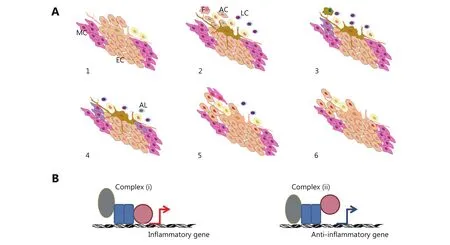
Figure 1 (A) Tissue response to internal or external insult followed by the activation of different cell types based on experimental evidence. The different cell types activated include several types of leukocytes (LC), resident macrophages (F), groups of resident cells in apoptosis (AC), and circulating components of the immune system migrating in successive waves (1-3). Selected groups of resident cells, such as epithelial cells (EC), undergo dedifferentiation into mesenchymal cells (MC) and re-epithelialization. Several mediators of immunosuppression are expressed. The immune response subsides, some leukocytes undergo apoptotic death (AL), and tissue function resumes (4-6). Accumulating data from molecular studies suggest that this succession of events is regulated by a chain of changes in the activity of sequence-specific transcription factors. In particular, factors that interact with Rel family proteins alter the gene selection activated by the Rel-domain proteins. This occurrence results in cell-specific patterns of gene expression and determines the function of each cell type during inflammation. (B) Simplified view of transcriptional regulation. Normally, transcription factor complex (i) is produced during inflammation, whereas complex (ii) appears during inflammatory resolution. After complex (ii) resolves, chromatin returns to its basal state, and the cell expresses genes depending on its typical function in the tissue. The cell is then prepared to induce complex (i) formation upon inflammatory stimulation. In cancer, pro- and anti-inflammatory mechanisms may be activated. The cancer cell may survive this occurrence by increasing the activity of intracellular turnover systems. Under the simultaneous expression of pro- and anti-inflammatory genes, a cancer cell is endowed with survival mechanisms under inflammation and sends mixed signals to its microenvironment.
bZip can form numerous potential dimers, where each monomer may be activated by discrete members of the mitogen-activated protein kinases. These kinases include the extracellular signal-activated kinases (ERK) and the stressactivated protein kinases [p38 and c-Jun kinase (JNK)], which promote cell apoptosis41. A tumor may activate an appropriate survival mechanism, such as degrading a key apoptotic mediator. In this case, NF-κB-induced inflammatory genes may facilitate metastasis by permitting cancer cells to overcome tissue barriers to invasion and express molecules that enable cancer cell adaptation in the metastatic niche42,43.
Cells may be stimulated by signals that induce later stages of inflammation, as well as ensuing tissue regeneration. In this case, NF-κB cooperates with transcription factors, such as STAT3, to induce the expression of genes that encode proteins mediating cellular transdifferentiation, resident stem-cell proliferation, immune cell apoptosis, and tissue protection from the immune system44. These proteins collectively operate to enable a tissue to return to a functional state. A cell will undergo apoptosis or survive depending on whether proteins that interact with the Bcl-2 family are expressed. NF-κB drives the expression of several antiapoptotic genes of the Bcl-2 family45. Cell stress can induce the NF-κB-dependent expression of protein p62, which sequesters proteins to the proteasome and lysosome, to clear protein aggregates25. In cancer, altered pace and selection of protein removal enables malignant cells to neutralize attacks from the immune system and consequently survive in niches that would normally reject the differentiated progeny of their parental cell lineage5,37,46. A comprehensive list of genes regulated by NF-κB is providedby the Gilmore Lab at http://www.bu.edu/nf-kb/generesources/target-genes/

Figure 2 Interaction between transcription factors NF-κB and AP-1, which bind to their respective DNA target sequences and facilitate chromatin remodeling. In the nucleus, NF-κB binds to cognate sites and synergistically interacts with proteins to redistribute chromatin remodeling complexes on chromosomal DNA. This change affects the cellular developmental fate by altering protein occupancy in the proximal and distal gene regulatory regions in a cell-specific and gene-specific fashion.
Operating mode of NF-κB
Inflammation activates successive waves of inducible transcription factors that enter the nucleus and bind to specific DNA sequences to modulate gene expression. Inflammation is elicited by the molecules released by dying cells or expressed on parasite surfaces. These molecules bind to specific cell surface transmembrane receptors, such as the TLR family or the RAGE, which in turn activate the expression and release of cytokines, such as TNF, IL-1, or CCL247-51. The released cytokines then stimulate other cells to express additional cytokines and surface receptors until adequate combinations of feedback repressors for inflammation are produced. At this point, tissue function resumes52. Although some transcription factors share few functions within a given gene cohort, the interactions among NF-κB and proteins of the AP-1 and STAT families have been the most studied operating system for inflammatory gene promoters19,23,53,54.
In most cells, the transcription factors of the NF-κB family appear in latent cytoplasmic forms. Under signals that induce either the phosphorylation or proteolysis of their inhibitor IκB, the transcription factors enter the nucleus almost instantly, whereas the most potent Rel protein, RelA (p65), which is phosphorylated on the Serine 536 residue, induces the expression of the κIBα gene to produce a timely negative feedback to NF-κB. Meanwhile, the expression of inflammatory genes depends on oxidative stress1. In this manner, gene expression induced by reactive oxygen species (ROS) is rapidly restrained by IκBα resynthesis. RelA serine 536 phosphorylation occurs mainly from the IKK complex and possibly through the contribution of other enzymes, such as protein kinase C isoforms delta and zeta55-57. Increased oxidative stress then induces RelA phosphorylation on serine 276 and concurrently preconditions the chromatin of inflammatory genes for NF-κB binding. Specifically, ROS promotes oxidative modifications on DNA, such as 7,8-dihydro-8-oxoguanine (8-oxoG), which induces the 8-oxoguanine DNA glycosylase1 (OGG1)-initiated base excision repair pathway. OGG1 strengthens NF-κB binding on DNA58. NF-κB activity on inflammatory genes is hence limited, and depends on ROS and the produced 8-oxoG base lesions. The activities of other transcription factors are then stimulated, and these factors can interact with NF-κB and modulate the expression of common target genes.
In particular, the c-Jun factor of the AP-1 family is transcribed and translated rapidly upon phosphorylation of factors that activate its own promoter59. As a result, the c-Jun protein becomes available to regulate transcription. AP-1 recruits the chromatin remodeling complex SWI/SNF60. STAT proteins can then be activated by JAK family kinases to fine tune the time course of inflammation in a tissue61. STAT proteins help recruit histone acetyltransferases through NF-κB62. Changes in the acetyltransferase abundance on cellfunction-related chromatin elements are critical for cell fate. For example, STAT3 increases the abundance of p300 on super-enhancers of genes characteristic to T-helper type 17 (Th17) cell function; this increase marks the genes critical for this cell type63.
The interactions among NF-κB, AP-1, and STAT proteins depend on the expression of different proportions of their subunits in different cell types. Stimulus-induced posttranslational modifications enable rapid inflammatory control. In primary epithelial cells, oxidized palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine activates tyrosine kinase JAK2, which in turn phosphorylates STAT3 on residue 705. This series of activations enables STAT3 binding to the sequence flanking the consensus γ-interferon activation sequence (GAS) in the promoter of the gene IL-8 encoding the cytokine IL-8/CXCL864. IL-8 binds to cognate transmembrane receptors that activate the chemotactic movement of neutrophils and monocytes across concentration gradients. Therefore, IL-8 significantlyinfluences local tissue inflammation65. However, the prolonged stimulation of immortalized cells by cytokine IL-6 can increase the expression of STAT3 without JAK2-catalyzed phosphorylation. STAT3 then activates the expression of the cytokine CCL5 by interacting with the p65 NF-κB bound to its target site on the gene promoter66.
Immortalized untransformed cells differ from carcinoma in protein kinase C activity. During cancer progression, protein kinase C may be activated by the tumor promoting agent phorbol 12-myristate 13-acetate (PMA)67,68. In a different fashion, stimulating epithelial carcinoma cells with cytokine IL-1 and not IL-6 may lead to the tethering of STAT3 without JAK2-catalyzed phosphorylation. This tethering is achieved through the interaction of STAT3 with the p65 NF-κB bound to its target site on gene promoters that activate the expression of IL-8/CXCL8 and other inflammatory genes69. Independently from synergy with IKK-induced NF-κB, starved cancer cells, activating ROS, autophagy, and kinase Rac1, enable unphosphorylated STAT3 to activate IL6 (IL-6) gene expression in synergy with NF-κB and independently from IKK70. Meanwhile, autophagy can activate the p65 phosphorylation on serine 536 by protein kinase C zeta independently from IKK56.
For feedback inhibitors, genes that encode inflammationrelated proteins can be divided into those that depend on RelA (p65) phosphorylation on residue 276 or residue 53623. The former genes encode potent inflammatory initiators, which are induced by oxidative stress and repressed by IκBα. Thus, these initiators are switched off by antioxidants and glucocorticoids71. By contrast, the latter genes encode proteins that modulate inflammation in a cell-specific manner. The presence of acetylases is critical for the genes driven by RelA phosphorylation on residue 536. The synergy of proteins, such as STAT3, facilitates the expression of genes that ultimately terminate immune response72. STAT3 interacts with multiple proteins that respond to organelle dynamic changes and consequently helps optimally regulate tissue postnatal development73,74. However, this optimum function requires strict limits on acetylase activity61. Tissues are protected from uncontrolled inflammation by synthesizing inhibitors, including IκBα75.
Protein complexes involved in the coordination between DNA repair and cell survival influence the regulation of NF-κB activity. Upon oncogene activation, tumor suppressor ARF induces the ATR- and Chk1-dependent phosphorylation of the RelA transactivation domain at threonine 505 that represses cell survival gene expression76. Furthermore, this phosphorylation on residue threonine 505 of RelA transforms NF-κB into an apoptotic gene activator and autophagy inhibitor. These functions are essential to limiting cell proliferation and migration and helps balance the supply of cellular components for tissue regeneration77. Consistently, knock-in mice with RelA T505A mutation develop normally but exhibit aberrant hepatocyte proliferation following liver partial hepatectomy or carbon tetrachloride (CCl4) induced damage. The role of T505 as an oncogenic inhibitor was then confirmed when the RelA T505A mice exhibited early cancer onset in the N-nitrosodiethylamine model of hepatocellular carcinoma78.
NF-κB activity can be steered also by nuclear receptors that are activated by metabolic products. For example, proteins of the peroxisome proliferator-activated receptor (PPAR) family are activated by free fatty acids, affect NF-κB subunit phosphorylation, and can promote NF-κB-driven apoptosis79,80. However, PPAR proteins possess multiple interaction partners in a cell, and their effects on cancer cells depend on many factors, including the cell’s metabolic activity81. Hence, PPAR activity may become either oncogenic or oncosuppressive82,83.
Loss of feedback to NF-κB affects tissue homeostatic coordination
Hypoxia normally limits NF-κB activity when NF-κB induces the expression of hypoxia-induced factor 1 (HIF1), which restricts the expression of NF-κB target genes, under physiological conditions. This process contributes to the regulation of extracellular milieu84,85. However, in contrast to normal pancreatic tissues or nontumorigenic cell lines, NF-κB is constitutively activated in most (>70%) human pancreatic cancer cells and primary tumor specimens. Molecular analysis shows a p65-HIF1 cooperation that drives epithelial-mesenchymal transition (EMT) and resistance to chemotherapy86. The analysis shows that N-cadherin, vimentin, Snail, and Twist overexpression, as well as E-cadherin suppression, induces striking morphological changes to loss of cell polarity, acquisition of mesenchymal phenotype, and resistance to gemcitabine.
Aberrant NF-κB activity bestows cancer cells with the capacity to overrun the natural control of cell bioenergetic and survival functions that normally depend on cell-specific interactions with the host tissue. Specifically, NF-κB simultaneously controls many molecules involved in cell communication with the rest of the organism. Interfering with NF-κB regulation is thus critical for virtually any type of aggressive cancer; this capacity enables unrestricted movement and survival in the presence of apoptotic inducers32. Unrestricted movement becomes possible bysynthesizing chemotactic mediators, such as IL-8, producing enzymes that break down molecules that restrict movement, and inactivating pathways that cause cell death in an aberrant niche5. Every category of these NF-κB target genes has multiple functions that simultaneously control many aspects of the activity they regulate in a cell-specific and coordinated fashion87,88.
Cell specificity is attained through the differences in the proportions of cytokine receptors, as well as the specific assortments of intracellular signal transducers89-91. The persistence of both chronic inflammation and cancer requires loss of feedback to NF-κB. However, in cancer, the loss of feedback allows independence from tissue phenotype control and enables cancer cells to grow in an aberrant niche microenvironment5. The tissue control normally limits the phenotype change possible for a cell; this control functions through the regulation of cell death, immune recognition of cell surface antigens, and effects of secreted cytokines on the signals that control stemness/differentiation of a cellular lineage92-95.
Surface antigen changes indicate that cancer cells may increase the expression of inflammatory mediators and enzymes that remodel the extracellular matrix without becoming a target for the immune system. Enhanced permeability would help activate immune response; however, cancer cells are not recognized by the immune system in a manner that allows effective tumor clearance.
Interacting agents of NF-κB direct immune system activity
The ultimate barrier to carcinogenesis is the immune system. The effects of the immune system depend heavily on the precise cell communication with all the components of the innate and adaptive immunity96. Defects in the cellular response to cytokines and adhesion molecules deprive the immune system of essential guidance in resolving infections and eliminating life-threatening developmental aberrations in host tissue cells97.
Changes are observed after oncogene activation in animal models. These changes suggest that cancer develops aberrant signals that suppress antitumor immunity on the basis of the normal signals that regulate inflammation and the ensuing partial tissue regeneration. Genes induced in cohorts during inflammation mediate the propagation and, ultimately, the termination of inflammatory processes to restore and protect host tissue. In mammals, two main cancer types occur, as follows: the solid tumor, which requires disruption of tissue adherence to metastasize, and the immune cell tumor, which readily forms under few molecular aberrations5. Immune cells of the myeloid lineage readily proliferate under innate immune response triggers. These cells possess redundant mechanisms for organelle turnover and NF-κB activation. By contrast, lymphoid cells can sustain survival and proliferation only under precise development and functional fidelity of the modules for acquired immunity98. Consequently, lymphoblastic leukemia develops NF-κB-driven chemoresistance far less often than myeloid leukemia99,100.
Inflammation through NF-κB interferes with postnatal tissue development and function by altering the activities of multiple enzymes in the nucleus. Macroscopically, inflammation interferes with tissue function101. Microscopically, groups of cells either die or reprogram their functions. Toward the end of inflammation, dead cells are replaced by reprogrammed resident cells, as well as by circulating cells that possess compatible differentiation capacity. At the molecular level, this interference between inflammation and tissue development becomes possible through shared cofactors between NF-κB and nuclear receptors that mediate the hormonal control of cell function within the tissue102. Cofactors of this type include zinc-finger LIM-domain proteins103,104.
The critical role of NF-κB regulation is evident in the control of the interaction between the cells of the monocytemacrophage lineage and the circulating T-cells in the bone marrow. T-cells secrete the receptor activator of NF-κB ligand (RANKL), which induces osteoblasts to differentiate into the monocytic lineage and give rise to osteoclasts that contribute to bone remodeling. Deregulation of RANKL can cause bone loss105.
In certain cancer cell categories, overexpressing proteins of the AP-1 and STAT families increases the recruitment of acetylases and impedes the function of the negative feedback loop on NF-κB. This occurrence then redirects the NF-κB activity on immunosuppressive genes106. In agreement with this effect, high STAT3 abundance results in a statistically increased risk of poor cancer prognosis107. The same is true for the combined NF-κB/STAT3 target gene that encodes cytokine IL-6108. STAT3 is critical for the function of superenhancers that regulate switching between Th1-, Th2-, and Th17-driven immunity63.
STAT3 and NF-κB enable macrophages to secrete the ligand PD-L1 that inhibits T-cell activity by binding to their PD-1 surface receptor109. NF-κB-driven PD-L1 expression is increased after stimulating cells with TNFα, TGFβ, or EGF, or constitutive activation of the EGFR that in cancer can accompany EMT, and increased vascular permeability; PDL1 apparently protects cancer cells that enter circulation andremain in circulation110-112. Interestingly, tumor-associated macrophages can capture therapeutic monoclonal antibodies raised against PD-1 from T-cell surfaces. This observation demonstrates the effect of macrophages on acquired immunity113. Macrophages can promote tumor evasion of immune response through multiple mechanisms that affect T-cell activity.
T-cells and macrophages are two key cell categories involved in immunity and respond promptly to cytokine changes114. T-helper cells secrete cytokine cocktails that direct acquired immunity. Macrophages, which are abundant in the organism, bind to microbial and other disease-related antigens and secrete more cytokines to recruit neutrophils, while presenting antigen to facilitate cooperation between cells of innate and adaptive immunity115. The critical role of the interaction between the cells of the monocytemacrophage lineage and the circulating T-cells is demonstrated in the bone marrow. T-cells secrete RANKL, which induces osteoblasts to differentiate into the monocytic lineage, giving rise to osteoclasts. Besides loss of bone, the formation of an effective premetastatic niche may occur in the bone marrow because of RANKL deregulation. This effect is achieved because the osteoclast is inefficient in antigen presentation105.
For the cytokine modulation of T-cell activity, several viruses secrete homologs of human cytokines such as IL-10, to impede antiviral immunity116. The limits of cytokine expression in macrophages and T-cells, are largely dependent on the regulation of NF-κB75,117. NF-κB is also involved in the expression of the anti-inflammatory glycoprotein MUC1 by epithelial cells118. MUC1 induction shows how inflammatory stimuli in normal tissue help restrain excessive inflammation during infection.
To summarize, the organism uses the immune system to control the activity of tissue-residing cells and phenotypes of circulating cells. This control is mainly achieved by secreting specific cytokine combinations. These combinations change depending on the availability of feedback regulators of NF-κB in each cell. These feedback regulators not only control internal cell functions but also determine the cell interaction with the rest of the organism (a comprehensive list of proteins that interact with NF-κB is presented in http://www.bu.edu/nf-kb/physiological-mediators/ interacting-proteins/).
Intracellular and extracellular cancer processes
In cancer, different modules of protein turnover show excessive activity because of cell stress from mutational burden and the need for bioenergetic adaptation in cells that deviate from tissue function5,119. Depending on the cell growth state, neoplastic development stage, and malignant cell location in the metastatic or primary niche, several regulators of protein synthesis and degradation can operate beyond the limits of normal cells. Protein synthesis control pathways include the signal cascade PI3K/AKT/mTOR, which controls the activation of ribosomal subunits and polyribosome formation, to allow protein translation120. Growth factors FL, EGF, PDGF, and IGF-1, activate AKT signaling121,122. Cancer cells may activate AKT kinase, and ribosomal activity concurrently with NF-κB123-125. Interestingly, the androgen receptor in renal cell carcinoma can enhance AKT/NF-κB activity to induce CXCL5 expression and endothelial cell recruitment, which in turn facilitate metastasis126. The AKT/S6/NF-κB pathways also allow cytokine-independent growth and hence afford malignant cells with substantial plasticity127,128.
Malignant cells can grow independently from mTOR signaling and the classical NF-κB cofactor BRD4 by activating autophagy. For example, in acute myeloid leukemia (AML), treating with the BRD4 inhibitor JQ1 activates AMPK and hence autophagy in cell populations with leukemia-initiating capacity119. Conversely, AML cells with mutations on the cytokine FL receptor FLT3 tyrosine kinase domain gain resistance to tyrosine kinase inhibitors through BRD4129.
Cytokine-independent growth is important because it allows a cancer cell to deviate from the extracellular signaling code of a tissue by secreting cytokines at time points and concentrations unrelated to normal tissue needs. Specifically, the range of effects enabled by this deviant secretion allows cancer cells to modulate their microenvironment, which includes fibroblasts and endothelial cells. As a result, stromal cell function is modified in both cancer primary and metastatic niche126,130-132. NF-κB activates the expression of metalloproteases, as well as chemokines, which enable the malignant cell to overcome tissue-specific restrictions in cellular motility133-135. Changes in the feedback-induced regulation of NF-κB activity allow cancer cells to activate several cytokines and modulators of cell development to recruit and alter cells of the monocyte/macrophage lineage136. Macrophages constitute a critical part of cytokinesecreting cells in a tissue, especially under low-oxygen tension and inflammation84. In pancreatic cancer, cultured M2-polarized macrophages significantly increase the migration rate of pancreatic ductal adenocarcinoma cells137.
Poor patient prognosis is possibly associated with the T-helper type 1 immune response (Th1) in non-small cell lungcancer (NSCLC). This relation is an example of extreme deviation from the regular cytokine code. Typically, the Th1 response is needed against tumors and viral infections. In the above-mentioned case, the T-cell-inactivating ligand PD-L1 on CD45+CD14+ monocytes/macrophages was more abundant in tumor tissue than in the adjacent nontumor tissues138. This observation can be reconciled with at least two possible scenarios. On one hand, inflammatory stimuli in the presence of glucocorticoids, TGFβ, and IL-10 can generate macrophages with an immunosuppressive M2-profile136.
However, the tumor cells may secrete PD-L1 themselves, especially when stimulated by PI3K/AKT activators (e.g., insulin, IGF, niche-homing SDF1/CXCR4 signals) or by IL-6-induced STAT3 activation to undergo EMT139-142. The Th1 response allows EMT through the following observation. In NSCLC specimens, discrete zones of Th2, Th17, and Th1 were found. These zones distinguished the tumor nest from the tumor boundary, adjacent normal lung tissue, or corresponding lymph node tissue138. IL-17 activates STAT3 and induces EMT in lung cancer cells in vitro. IL-17 also correlates with EMT in human lung cancer specimens143. TNFα and TGFβ enhance p65-driven expression of Twist1 and the self-renewal of “cancer stem cells (CSC), ” which are malignant clones with established capacity to initiate tumors144. EMT-activated p65 can induce the expression of the TGFβ-family member Activin, which in turn induces EMT master-switch regulators and self-renewal factors that can sustain CSC and enable metastasis145. The cytokines secreted in the vicinity of cancer cells can thus permit the propagation of clones that function as CSCs through the positive feedback activation of EMT.
On one hand, these defects in feedback restriction of NF-κB activity allow proliferating cancer cells to survive. On the other hand, these defects cause tumor stroma and tumor nest to interact aberrantly with the immune system and cripple the immune response within the tumor microenvironment. In other words, several control molecules for the immune response can establish immunosuppressive niches for the cancer cells, which would then be difficult to detect macroscopically.
Modulation of the microenvironment to “license” cancer: many processes initiated by a few genes
Proteins directly regulated by NF-κB at the transcriptional level include many NF-κB transcriptional cofactors. Several of these proteins are also regulated by STAT3, as well as by STAT3 synergy with NF-κB depending on the cell state. Such important proteins can be encountered on the chromatin of the chemokine IL-8, which facilitates leukocyte and endothelial cell movement and reprogramming. IL-8 also contributes to inflammation-induced tissue remodeling and vascularization146. Notably, IL-8 does not exert ubiquitous effects in all cell types. For instance, it cannot initiate extracellular matrix remodeling in the nucleus pulposus cells of the human intervertebral disc147.
The interaction of NF-κB with the il8 gene promoter can integrate regulation through different types of transcription factors, including AP-1, EGR1, helicase WRN, STAT3, and MUC116,64,148-151. β-catenin also mediates IL8 gene expression after receiving signals that activate WNT or TLR4152,153. TLR4 activates phosphorylases p38 and ERK154. p38 and ERK activate MSK1, which phosphorylates RelA on Ser276155. TLR4 by activating p38, induces NF-κB to bind chromatin DNA in the nucleus156. NF-κB recruits bromodomain proteins such as BRD4, which enable chromatin remodeling in synergy with acetyltransferases p300/CBP, which are recruited by the above-mentioned transcription factors157. AP-1 helps recruit the SWI/SNF chromatin remodeler. However, this remodeler requires target specificity in BRD460,158. Therefore at least in myeloid cells, AP-1 acts as an amplifier of NF-κB-driven IL8 gene expression16.
NF-κB also recruits BRD4 to the genomic regulatory region of Myc27. In leukemia cells, BRD4 and SWI/SNF maintain transcription factor occupancy on the Myc gene and facilitate promoter interactions with a lineage-specific super-enhancer located 1.7 megabases at the 3’ direction from the Myc gene159. However, the product c-Myc does not participate in il8 gene regulation like the other NF-κB targets160. The genomic locus of Myc contains abundant STAT3 DNA binding sites. This observation is consistent with the expression of Myc in cells “licensed” to proliferate5. In cancer, c-Myc activates the expression of several metabolic enzymes that enable cell proliferation. c-Myc also requires NF-κB activity for cell survival, because Myc induces the expression of apoptotic genes161.
Besides being a transcriptional cofactor, MUC1 is encoded by an NF-κB target gene (Figure 3). The MUC1 protein regulates tissue metabolism, integrity, and gene expression162. MUC1-C/β-catenin/TCF4 complexes promote p300 recruitment on the Myc promoter. This observation indicates that β-catenin-complexes can induce Myc expression in response to BRD4-dependent inflammatory signals163. MUC1-C complexed with NF-κB activates the expression of enzyme DNMT1, which methylates and inactivates tumor suppressor genes and then permits malignant cell growth over time164.

Figure 3 Genomic locus of the MUC1 gene on human chromosome 1. (A) The locus contains DNA binding sites for the RelA subunit of NF-κB. (B) NF-κB binding sites increase in abundance when transcription factors that interact with NF-κB bind to their cognate DNA target sites, and thereby recruit NF-κB to the locus. These transcription factors change in activity during inflammation. The product of the MUC1 gene is the protein MUC1, which participates in cytokine gene regulation and tissue homeostasis.
The full-length product of the MUC1 gene, MUC1, is a transmembrane protein normally expressed on the luminal surfaces of ductal epithelia. MUC1 regulates apical-basal polarity and fine tunes macrophage phenotypes, whereas the MUC1 protein-derived cytoplasmic domain provides feedback regulation to NF-κB transcriptional activity165,166. MUC1 can interact with β-catenin and p120 catenin to modulate WNT signaling. That is, in pancreatic cancer cells, MUC1 augments the activity of cyclin D1 gene (CCND1) promoter through β-catenin and TCF-Lef, and by stabilizing p120 catenin isoforms that displace the repressor protein KAISO167. The cytoplasmic domain of MUC1 (MUC1-C) spurs cell growth by recruiting β-catenin and acetylase p300 on the genes CCND1 and Myc168. In breast cancer cells, the complexes of MUC1-C/STAT3 can be also detected on the promoters of CCND1 and MUC1, which are STAT3 target genes169. T helper (Th) 17 cells produce the effector cytokine IL-17 along with IL-22, which stimulates colonic epithelial cells to produce MUC1. MUC1 normally switches off Th17-driven inflammation170. However, in neoplasia, MUC1-C and STAT3 link cytokine-induced inflammatory response to cancer cell survival.
MUC1-C interacts directly with RelA at the Rel homology domain (RHD). MUC1 blocks binding of RelA to IκBα, and thereby releases NF-κB169. In carcinoma cells, MUC1-C generally provides positive feedback to the STAT1/3 and NF-κB RelA complex that activates the MUC1 gene171. Experiments on pancreatic cancer cells showed that MUC1 stabilizes lysosomes, which are an important turnover system and an auxiliary system for NF-κB induction after cell stress, sparked by inhibition of proteasome activity24,172,173. Through NF-κB, β-catenin and MUC1, lung cancer cells interact with M2-polarized macrophages and gain stemness properties174. Therefore, muc1 is a characteristic target gene of NF-κB that could provide cancer cell populations with vital transcriptional regulation and concurrently affect the microenvironment of a tumor nest.
NF-κB target gene regulation is tissue dependent
Chromatin remodeling complexes act in a highly locus-specific and cell-specific manner. Notably, the SWI/SNF subunit Brg-1 activates tumor-suppressing mechanisms in solid tumors, as well as potent oncogenes in leukemias159,175,176. This contrast can be explained by the parallel role of some molecules involved in extracellular activity in regulating gene expression. NF-κB activity modulators drive changes in cellular programming, while modulating tissue interactions and the immune response5. These changes are evident in the altered abundance of chromatin-modifying molecules on key regulatory regions of the chromosomal DNA that controls cell activity7. These alterations manifest in the cells that steer the immune response and result in measurable changes in cytokine abundance. In cancer, a part of this regulatory link between the intracellular control of cell proliferation and extracellular activity fails to function accurately177. At the cellular level, interaction with the rest of the organism involves diverse molecular types. However, the changes detectable by basic methodology typically involve cytokines101. Cytokine abundance changes in pathological conditions that involve inflammation.
Yet in cancer, aberrant activity of the genes involved in cytokine feedback regulation in tissues provides malignant cells with amplifying signals for cell proliferation and metastasis. This occurrence is expected to critically influence the disease course177. In solid tumors NF-κB overexpression is generally linked to worsened overall survival at 3, 5, or 10 years after diagnosis178. However, the molecular aberrations are not uniform, even within a given cancer type. These variations thereby complicate the epidemiological analysis of systemic cytokine levels. Higher serum IL-6 levels correlate with poor prognosis, but the degree of epidemiological association with prognosis varies across studies108. In NSCLC, high IL-6 concentration is associated with poor prognosis in patients treated with chemotherapy but not in those who underwent surgery179. When considering transcription factors, high STAT3 expression marks poor prognosis in NSCLC180. STAT3-activating cytokines IL-6, IL-10, and IL-17 do not show a uniform prognostic profile in lung cancer181. This complexity can be attributed to the distinct homeostatic roles of each cytokine in a given tissue context. Cancer cells deviate in their response to cytokine stimulation depending on the composition of their internal regulatory network, and this deviation is linked to the developmental cell fate.
A different example of a network's context dependence between cell genomic integrity and the immune system is triple-negative breast cancer. BRCA1 is a DNA repair protein functioning as a cofactor for the expression of the RelA-target DNMT1 (involved in the long-term regulation of gene expression). BRCA1 apparently determines the immune response switching from oncosuppressive Th1 to oncogenic Th2182,183. BRCA1 loss allows NF-κB-driven immune signals to maintain the M1 macrophage, which supports the sustained presence of cytotoxic CD8+ T-cells. In this case, deficiency of the DNA repair protein BRCA1 switches the tumor microenvironment to an oncosuppressive mode associated with a favorable disease prognosis. Evidently the cooperation between resident macrophages and infiltrating T-cells is critical in the cells’ individual functions within the tumor niche.
Conclusions
Inflammation is a process that challenges the developmental integrity of a tissue. This process activates mechanisms that disrupt optimum hormonal function and interfere with the coordination of gene expression between different cellular components. Apoptotic mechanisms tie normal cell survival with functional fidelity within the organism. The transcription factor NF-κB is regulated by key modules of protein turnover. The transcription factor acts as a mediator for apoptotic and survival signals. NF-κB integrates the input from signaling cascades that alter post-translational modifications of NF-κB subunits, as well as the input from the feedback regulation exerted by the protein products of its own gene targets. Malignant cells acquire defects in the coordination of intracellular turnover with the extracellular cell communication, with the harboring tissue, and the response of the cell to circulating hormonal mediators. Some of these defects interfere with the regulation of the cellular response to NF-κB activity induction, reflected in the coordination of genomic function. As a result, the disrupting of NF-κB feedback control by its target genes changes the intensity and duration of inflammatory gene expression in the cells of the tumor nest, and their microenvironment. This change enables cancer cells to escape immune surveillance and grow in metastatic niches. Dissecting the gene regulation dynamics in the tumor nest and tumor stroma would yield insights in the capacity of cells to migrate and interact within the metastatic niche.
Acknowledgements
Figure 3 used an illustration derived from the Human Genome Browser. The University of California at Santa Cruz project team for the Human Genome Browser (assembly hg19) is composed of Hiram Clawson, Brooke Rhead,Pauline Fujita, Ann Zweig, Katrina Learned, Donna Karolchik, and Robert Kuhn (website: http://genome. ucsc.edu/goldenPath/credits.html#human_credits)
Starting with the hg19 assembly, the human genome sequence was obtained from the Genome Reference Consortium (GRC). The credits for the GRC are listed in http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/ credits.shtml.
Conflict of interest statement
S. Vlahopoulos is listed as co-inventor in patent applications that describe the subcellular targeting of substances potentially interfering with autophagy and transcription factor activity in a tissue-selective mode.
1.Choudhary S, Boldogh I, Brasier AR. Inside-out signaling pathways from nuclear reactive oxygen species control pulmonary innate immunity. J Innate Immun. 2016; 8: 143-55.
2.Crivellaro S, Panuzzo C, Carrà G, Volpengo A, Crasto F, Gottardi E, et al. Non genomic loss of function of tumor suppressors in CML: BCR-ABL promotes IκBα mediated p53 nuclear exclusion. Oncotarget. 2015; 6: 25217-25.
3.Pazarentzos E, Mahul-Mellier AL, Datler C, Chaisaklert W, Hwang MS, Kroon J, et al. IκΒα inhibits apoptosis at the outer mitochondrial membrane independently of NF-κB retention. EMBO J. 2014; 33: 2814-28.
4.Cogswell PC, Kashatus DF, Keifer JA, Guttridge DC, Reuther JY, Bristow C, et al. NF-κB and IκBα are found in the mitochondria. Evidence for regulation of mitochondrial gene expression by NF-κB. J Biol Chem. 2003; 278: 2963-8.
5.Vlahopoulos SA, Cen O, Hengen N, Agan J, Moschovi M, Critselis E, et al. Dynamic aberrant NF-κB spurs tumorigenesis: A new model encompassing the microenvironment. Cytokine Growth Factor Rev. 2015; 26: 389-403.
6.Sepuri NBV, Tammineni P, Mohammed F, Paripati A. Nuclear transcription factors in the mitochondria: a new paradigm in finetuning mitochondrial metabolism. In: Handbook of Experimental Pharmacology. Berlin Heidelberg: Springer; 2016. doi: 10.1007/164_2016_3.
7.Schmidt SF, Larsen BD, Loft A, Nielsen R, Madsen JGS, Mandrup S. Acute TNF-induced repression of cell identity genes is mediated by NFκB-directed redistribution of cofactors from superenhancers. Genome Res. 2015; 25: 1281-94.
8.Brown JD, Lin CY, Duan Q, Griffin G, Federation AJ, Paranal RM, et al. NF-κB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol Cell. 2014; 56: 219-31.
9.Kalita M, Tian B, Gao BN, Choudhary S, Wood TG, Carmical JR, et al. Systems approaches to modeling chronic mucosal inflammation. BioMed Res Int. 2013; 2013: 505864
10.Park MH, Hong JT. Roles of NF-κB in cancer and inflammatory diseases and their therapeutic approaches. Cells. 2016; 5: E15 doi: 10.3390/cells5020015.
11.Ba XQ, Aguilera-Aguirre L, Sur S, Boldogh I. 8-oxoguanine DNA glycosylase-1-driven DNA base excision repair: role in asthma pathogenesis. Curr Opin Allergy Clin Immunol. 2015; 15: 89-97.
12.Brasier AR. The NF-κB regulatory network. Cardiovasc Toxicol. 2006; 6: 111-30.
13.Mihalas AB, Meffert MK. IKK kinase assay for assessment of canonical NF-κB activation in neurons. In: May MJ, editor. NF-kappa B: Methods and Protocols. New York: Springer. 2015; 61-74.
14.Häcker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006; 2006: re13.
15.Zhao YX, Tian B, Edeh CB, Brasier AR. Quantitation of the dynamic profiles of the innate immune response using multiplex selected reaction monitoring-mass spectrometry. Mol Cell Proteomics. 2013; 12: 1513-29.
16.Vlahopoulos S, Boldogh I, Casola A, Brasier AR. Nuclear factorkappaB-dependent induction of interleukin-8 gene expression by tumor necrosis factor alpha: evidence for an antioxidant sensitive activating pathway distinct from nuclear translocation. Blood. 1999; 94: 1878-89.
17.Cieślik M, Bekiranov S. Genome-wide predictors of NF-κB recruitment and transcriptional activity. BioData Mining. 2015; 8: 37
18.Casola A, Garofalo RP, Jamaluddin M, Vlahopoulos S, Brasier AR. Requirement of a novel upstream response element in respiratory syncytial virus-induced IL-8 gene expression. J Immunol. 2000; 164: 5944-51.
19.Brasier AR, Jamaluddin M, Casola A, Duan WL, Shen Q, Garofalo RP. A promoter recruitment mechanism for tumor necrosis factor-α-induced interleukin-8 transcription in type II pulmonary epithelial cells. Dependence on nuclear abundance of Rel A, NF-κB1, and c-Rel transcription factors. J Biol Chem. 1998; 273: 3551-61.
20.Aguilera-Aguirre L, Bacsi A, Radak Z, Hazra TK, Mitra S, Sur S, et al. Innate inflammation induced by the 8-oxoguanine DNA glycosylase-1-KRAS-NF-κB pathway. J Immunol. 2014; 193: 4643-53.
21.Zhao WN, Breese E, Bowers A, Hoggatt J, Pelus LM, Broxmeyer HE, et al. SIMPL enhancement of tumor necrosis factor-α dependent p65-MED1 complex formation is required for mammalian hematopoietic stem and progenitor cell function. PLoS One. 2013; 8: e61123
22.Tian B, Nowak DE, Jamaluddin M, Wang SF, Brasier AR. Identification of direct genomic targets downstream of the nuclear factor-κB transcription factor mediating tumor necrosis factor signaling. J Biol Chem. 2005; 280: 17435-48.
23.Jamaluddin M, Wang SF, Boldogh I, Tian B, Brasier AR. TNF-αinduced NF-κB/RelA Ser276 phosphorylation and enhanceosome formation is mediated by an ROS-dependent PKAc pathway. CellSignal. 2007; 19: 1419-33.
24.Moschovi M, Critselis E, Cen O, Adamaki M, Lambrou GI, Chrousos GP, et al. Drugs acting on homeostasis: challenging cancer cell adaptation. Expert Rev Anticancer Ther. 2015; 15: 1405-17.
25.Song C, Mitter SK, Qi X, Beli E, Rao HV, Ding J, et al. Oxidative stress-mediated NFκB phosphorylation upregulates p62/SQSTM1 and promotes retinal pigmented epithelial cell survival through increased autophagy. PLoS One. 2017; 12: e0171940
26.Shaaban S, Negm A, Ibrahim EE, Elrazak AA. Chemotherapeutic agents for the treatment of hepatocellular carcinoma: efficacy and mode of action. Oncol Rev. 2014; 8: 246
27.Sun BH, Shah B, Fiskus W, Qi J, Rajapakshe K, Coarfa C, et al. Synergistic activity of BET protein antagonist-based combinations in mantle cell lymphoma cells sensitive or resistant to ibrutinib. Blood. 2015; 126: 1565-74.
28.Zhang BG, Hu L, Zang MD, Wang HX, Zhao W, Li JF, et al. Helicobacter pylori CagA induces tumor suppressor gene hypermethylation by upregulating DNMT1 via AKT-NFκB pathway in gastric cancer development. Oncotarget. 2016; 7: 9788-800.
29.Hu GK, Gong AY, Wang Y, Ma SB, Chen XQ, Chen J, et al. LincRNA-Cox2 promotes late inflammatory gene transcription in macrophages through modulating SWI/SNF-mediated chromatin remodeling. J Immunol. 2016; 196: 2799-808.
30.Jones VS, Huang RY, Chen LP, Chen ZS, Fu LW, Huang RP. Cytokines in cancer drug resistance: Cues to new therapeutic strategies. Biochim Biophys Acta. 2016; 1865: 255-65.
31.Renema N, Navet B, Heymann MF, Lezot F, Heymann D. RANKRANKL signalling in cancer. Biosci Rep. 2016; 36: e00366 doi: 10.1042/BSR20160150.
32.Dmitrieva OS, Shilovskiy IP, Khaitov MR, Grivennikov SI. Interleukins 1 and 6 as Main Mediators of Inflammation and Cancer. Biochem Biokhimiia. 2016; 81: 80-90.
33.Klein JC, Moses K, Zelinskyy G, Sody S, Buer J, Lang S, et al. Combined toll-like receptor 3/7/9 deficiency on host cells results in T-cell-dependent control of tumour growth. Nat Commun. 2017; 8: 14600
34.Park H, Kim SH, Cho YM, Ihm HJ, Oh YS, Hong SH, et al. Increased expression of nuclear factor kappa-B p65 subunit in adenomyosis. Obstet Gynecol Sci. 2016; 59: 123-9.
35.Haricharan S, Li Y. STAT signaling in mammary gland differentiation, cell survival and tumorigenesis. Mol Cell Endocrinol. 2014; 382: 560-9.
36.Park MH, Hong JE, Hwang CJ, Choi M, Choi JS, An YJ, et al. Synergistic inhibitory effect of cetuximab and tectochrysin on human colon cancer cell growth via inhibition of EGFR signal. Arch Pharm Res. 2016; 39: 721-9.
37.Yun HM, Park MH, Kim DH, Ahn YJ, Park KR, Kim TM, et al. Loss of presenilin 2 is associated with increased iPLA2 activity and lung tumor development. Oncogene. 2014; 33: 5193-200.
38.Qiao YC, He H, Jonsson P, Sinha I, Zhao CY, Dahlman-Wright K. AP-1 is a key regulator of proinflammatory cytokine TNFαmediated triple-negative breast cancer progression. J Biol Chem. 2016; 291: 5068-79.
39.Vlahopoulos SA, Logotheti S, Mikas D, Giarika A, Gorgoulis V, Zoumpourlis V. The role of ATF-2 in oncogenesis. BioEssays. 2008; 30: 314-27.
40.Bhoumik A, Takahashi S, Breitweiser W, Shiloh Y, Jones N, Ronai Z. ATM-dependent phosphorylation of ATF2 is required for the DNA damage response. Mol Cell. 2005; 18: 577-87.
41.Vlahopoulos S, Zoumpourlis VC. JNK: a key modulator of intracellular signaling. Biochemistry. 2004; 69: 844-54.
42.Park WY, Hong BJ, Lee J, Choi C, Kim MY. H3K27 demethylase JMJD3 employs the NF-κB and BMP signaling pathways to modulate the tumor microenvironment and promote melanoma progression and metastasis. Cancer Res. 2016; 76: 161-70.
43.Vasseur R, Skrypek N, Duchêne B, Renaud F, Martínez-Maqueda D, Vincent A, et al. The mucin MUC4 is a transcriptional and post-transcriptional target of K-ras oncogene in pancreatic cancer. Implication of MAPK/AP-1, NF-κB and RalB signaling pathways. Biochim Biophys Acta. 2015; 1849: 1375-84.
44.Yun HM, Jin P, Park KR, Hwang J, Jeong HS, Kim EC, et al. Thiacremonone potentiates anti-oxidant effects to improve memory dysfunction in an APP/PS1 transgenic mice model. Mol Neurobiol. 2016; 53: 2409-20.
45.Thompson RC, Vardinogiannis I, Gilmore TD. The sensitivity of diffuse large B-cell lymphoma cell lines to histone deacetylase inhibitor-induced apoptosis is modulated by BCL-2 family protein activity. PLoS One. 2013; 8: e62822
46.Lee NJ, Choi DY, Song JK, Jung YY, Kim DH, Kim TM, et al. Deficiency of C-C chemokine receptor 5 suppresses tumor development via inactivation of NF-κB and inhibition of monocyte chemoattractant protein-1 in urethane-induced lung tumor model. Carcinogenesis. 2012; 33: 2520-8.
47.Fernandez MV, Miller E, Krammer F, Gopal R, Greenbaum BD, Bhardwaj N. Ion efflux and influenza infection trigger NLRP3 inflammasome signaling in human dendritic cells. J Leukoc Biol. 2016; 99: 723-34.
48.Hosakote YM, Brasier AR, Casola A, Garofalo RP, Kurosky A. Respiratory Syncytial Virus infection triggers epithelial HMGB1 release as a damage-associated molecular pattern promoting a monocytic inflammatory response. J Virol. 2016; 90: 9618-31. doi: 10.1128/JVI.01279-16.
49.Kolli D, Gupta MR, Sbrana E, Velayutham TS, Chao H, Casola A, et al. Alveolar macrophages contribute to the pathogenesis of human metapneumovirus infection while protecting against respiratory syncytial virus infection. Am J Respir Cell Mol Biol. 2014; 51: 502-15.
50.Mastellos DC, DeAngelis RA, Lambris JD. Complement-triggered pathways orchestrate regenerative responses throughout phylogenesis. Semin Immunol. 2013; 25: 29-38.
51.Bai L, Chen W, Chen JT, Li W, Zhou L, Niu C, et al. Heterogeneity of Toll-like receptor 9 signaling in B cell malignancies and its potential therapeutic application. J Transl Med. 2017; 15: 51
52.Chen BJ, Frangogiannis NG. Immune cells in repair of the infarcted myocardium. Microcirculation. 2016; 24 doi: 10.1111/micc.12305.
53.Jamaluddin M, Choudhary S, Wang SF, Casola A, Huda R, Garofalo RP, et al. Respiratory syncytial virus-inducible BCL-3 expression antagonizes the STAT/IRF and NF-κB signaling pathways by inducing histone deacetylase 1 recruitment to the interleukin-8 promoter. J Virol. 2005; 79: 15302-13.
54.Ray S, Zhao YX, Jamaluddin M, Edeh CB, Lee C, Brasier AR. Inducible STAT3 NH2 terminal mono-ubiquitination promotes BRD4 complex formation to regulate apoptosis. Cell Signal. 2014; 26: 1445-55.
55.Bao XY, Indukuri H, Liu TS, Liao SL, Tian B, Brasier AR, et al. IKKε modulates RSV-induced NF-κB-dependent gene transcription. Virology. 2010; 408: 224-31.
56.Chamoux E, McManus S, Laberge G, Bisson M, Roux S. Involvement of kinase PKC-zeta in the p62/p62(P392L)-driven activation of NF-κB in human osteoclasts. Biochim Biophys Acta. 2013; 1832: 475-84.
57.Ren J, Wang QW, Morgan S, Si Y, Ravichander A, Dou CL, et al. Protein kinase C-δ (PKCδ) regulates proinflammatory chemokine expression through cytosolic interaction with the NF-κB subunit p65 in vascular smooth muscle cells. J Biol Chem. 2014; 289: 9013-26.
58.Pan L, Zhu B, Hao WJ, Zeng XL, Vlahopoulos SA, Hazra TK, et al. Oxidized guanine base lesions function in 8-oxoguanine DNA glycosylase1-mediated epigenetic regulation of nuclear factor κB-driven gene expression. J Biol Chem. 2016; 291: 25553-6. doi: 10.1074/jbc.M116.751453.
59.Mina M, Magi S, Jurman G, Itoh M, Kawaji H, Lassmann T, et al. Promoter-level expression clustering identifies time development of transcriptional regulatory cascades initiated by ErbB receptors in breast cancer cells. Sci Rep. 2015; 5: 11999
60.Yaniv M. Chromatin remodeling: from transcription to cancer. Cancer Genet. 2014; 207: 352-7.
61.Zouein FA, Altara R, Chen Q, Lesnefsky EJ, Kurdi M, Booz GW. Pivotal importance of STAT3 in protecting the heart from acute and chronic stress: new advancement and unresolved issues. Front Cardiovasc Med. 2015; 2: 36
62.Kamitani S, Togi S, Ikeda O, Nakasuji M, Sakauchi A, Sekine Y, et al. Krüppel-associated box-associated protein 1 negatively regulates TNF-α-induced NF-κB transcriptional activity by influencing the interactions among STAT3, p300, and NF-κB/p65. J Immunol. 2011; 187: 2476-83.
63.Witte S, Bradley A, Enright AJ, Muljo SA. High-density P300 enhancers control cell state transitions. BMC Genomics. 2015; 16: 903
64.Gharavi NM, Alva JA, Mouillesseaux KP, Lai C, Yeh M, Yeung W, et al. Role of the Jak/STAT pathway in the regulation of interleukin-8 transcription by oxidized phospholipids in vitro and in atherosclerosis in vivo. J Biol Chem. 2007; 282: 31460-8.
65.Sakelliou A, Fatouros IG, Athanailidis I, Tsoukas D, Chatzinikolaou A, Draganidis D, et al. Evidence of a redoxdependent regulation of immune responses to exercise-induced inflammation. Oxid Med Cell Longev. 2016; 2016: 2840643
66.Yang JB, Liao XD, Agarwal MK, Barnes L, Auron PE, Stark GR. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFκB. Genes Dev. 2007; 21: 1396-408.
67.Dong L, Stevens JL, Jaken S. Transformation-sensitive localization of alpha-protein kinase C at cell-cell contacts in rat renal proximal tubule epithelial cells. Cell Growth Differ. 1993; 4: 793-8.
68.Li J, Vesey DA, Johnson DW, Gobe GC. Erythropoietin reduces cisplatin-induced apoptosis in renal carcinoma cells via a PKC dependent pathway. Cancer Biol Ther. 2007; 6: 1944-50.
69.Yoshida Y, Kumar A, Koyama Y, Peng HB, Arman A, Boch JA, et al. Interleukin 1 activates STAT3/nuclear factor-κB cross-talk via a unique TRAF6- and p65-dependent mechanism. J Biol Chem. 2004; 279: 1768-76.
70.Kim SJ, Yoon S. Activated Rac1 regulates the degradation of IκBα and the nuclear translocation of STAT3-NFκB complexes in starved cancer cells. Exp Mol Med. 2016; 48: e231
71.Doucas V, Shi Y, Miyamoto S, West A, Verma I, Evans RM. Cytoplasmic catalytic subunit of protein kinase A mediates crossrepression by NF-kappa B and the glucocorticoid receptor. Proc Natl Acad Sci USA. 2000; 97: 11893-8.
72.Lee H, Herrmann A, Deng JH, Kujawski M, Niu GL, Li ZW, et al. Persistently activated Stat3 maintains constitutive NF-κB activity in tumors. Cancer Cell. 2009; 15: 283-93.
73.Xu YS, Liang JJ, Wang YM, Zhao XZJ, Xu L, Xu YY, et al. STAT3 undergoes acetylation-dependent mitochondrial translocation to regulate pyruvate metabolism. Sci Rep. 2016; 6: 39517
74.Walker EC, Johnson RW, Hu YF, Brennan HJ, Poulton IJ, Zhang J-G, et al. Murine oncostatin m acts via leukemia inhibitory factor receptor to phosphorylate signal transducer and activator of transcription 3 (STAT3) but not STAT1, an Effect that protects bone mass. J Biol Chem. 2016; 291: 21703-16.
75.Chatterjee B, Banoth B, Mukherjee T, Taye N, Vijayaragavan B, Chattopadhyay S, et al. Late-phase synthesis of IκBα insulates the TLR4-activated canonical NF-κB pathway from noncanonical NF-κB signaling in macrophages. Sci Signal. 2016; 9: ra120
76.Rocha S, Garrett MD, Campbell KJ, Schumm K, Perkins ND. Regulation of NF-κB and p53 through activation of ATR and Chk1 by the ARF tumour suppressor. EMBO J. 2005; 24: 1157-69.
77.Christian F, Smith EL, Carmody RJ. The regulation of NF-κB subunits by phosphorylation. Cells. 2016; 5: 12 doi: 10.3390/cells5010012.
78.Moles A, Butterworth JA, Sanchez A, Hunter JE, Leslie J, Sellier H, et al. A RelA(p65) Thr505 phospho-site mutation reveals an important mechanism regulating NF-κB-dependent liver regeneration and cancer. Oncogene. 2016; 35: 4623-32.
79.Mangoni M, Sottili M, Gerini C, Desideri I, Bastida C, Pallotta S, et al. A PPAR-gamma agonist protects from radiation-induced intestinal toxicity. United Eur Gastroenterol J. 2017; 5: 218-26.
80.Hwang JW, Cho H, Lee JY, Jeon Y, Kim SN, Lee SJ, et al. The synthetic ajoene analog SPA3015 induces apoptotic cell deaththrough crosstalk between NF-κB and PPARγ in multidrugresistant cancer cells. Food Chem Toxicol. 2016; 96: 35-42.
81.Pazienza V, Vinciguerra M, Mazzoccoli G. PPARs signaling and cancer in the gastrointestinal system. PPAR Res. 2012; 2012: 560846
82.Pozzi A, Capdevila JH. PPARα ligands as antitumorigenic and antiangiogenic agents. PPAR Res. 2008; 2008: 906542
83.Narayanan KB, Ali M, Barclay BJ, Cheng Q, D’Abronzo L, Dornetshuber-Fleiss R, et al. Disruptive environmental chemicals and cellular mechanisms that confer resistance to cell death. Carcinogenesis. 2015; 36(Suppl 1): S89-110. doi: 10.1093/carcin/bgv032.
84.Fecher RA, Horwath MC, Friedrich D, Rupp J, Deepe GS Jr. Inverse correlation between IL-10 and HIF-1α in macrophages infected with Histoplasma capsulatum. J Immunol. 2016; 197: 565-79.
85.Ben-Shoshan J, Maysel-Auslender S, Mor A, Keren G, George J. Hypoxia controls CD4+CD25+ regulatory T-cell homeostasis via hypoxia-inducible factor-1α. Eur J Immunol. 2008; 38: 2412-8.
86.Cheng ZX, Wang DW, Liu T, Liu WX, Xia WB, Xu J, et al. Effects of the HIF-1α and NF-κB loop on epithelial-mesenchymal transition and chemoresistance induced by hypoxia in pancreatic cancer cells. Oncol Rep. 2014; 31: 1891-8.
87.Chen JC, Huang AJ, Chen SC, Wu JL, Wu WM, Chiang HS, et al. Interleukin-27 and interleukin-12 augment activation of distinct cord blood natural killer cells responses via STAT3 pathways. J Formos Med Assoc. 2012; 111: 275-83.
88.Ma XJ, Yan WJ, Zheng H, Du QL, Zhang LX, Ban Y, et al. Regulation of IL-10 and IL-12 production and function in macrophages and dendritic cells. F1000Res. 2015; 4: F1000 doi: 10.12688/f1000research.7010.1.
89.Larousserie F, Charlot P, Bardel E, Froger J, Kastelein RA, Devergne O. Differential effects of IL-27 on human B cell subsets. J Immunol. 2006; 176: 5890-7.
90.El-behi M, Ciric B, Yu S, Zhang GX, Fitzgerald DC, Rostami A. Differential effect of IL-27 on developing versus committed Th17 cells. J Immunol. 2009; 183: 4957-67.
91.Xu J, Yang Y, Qiu G, Lal G, Wu Z, Levy DE, et al. c-Maf regulates IL-10 expression during Th17 polarization. J Immunol. 2009; 182: 6226-36.
92.Prasongsukarn K, Chaisri U, Chartburus P, Wetchabut K, Benjathummarak S, Khachansaksumet V, et al. Phenotypic alterations in human saphenous vein culture induced by tumor necrosis factor-alpha and lipoproteins: a preliminary development of an initial atherosclerotic plaque model. Lipids Health Dis. 2013; 12: 132
93.Barone F, Gardner DH, Nayar S, Steinthal N, Buckley CD, Luther SA. Stromal fibroblasts in tertiary lymphoid structures: a novel target in chronic inflammation. Front Immunol. 2016; 7: 477
94.Milićević NM, Nohroudi K, Schmidt F, Schmidt H, Ringer C, Sorensen GL, et al. Growth of murine splenic tissue is suppressed by lymphotoxin β-receptor signaling (LTβR) originating from splenic and non-splenic tissues. PLoS One. 2016; 11: e0166901
95.Visciano C, Liotti F, Prevete N, Cali’ G, Franco R, Collina F, et al. Mast cells induce epithelial-to-mesenchymal transition and stem cell features in human thyroid cancer cells through an IL-8-Akt-Slug pathway. Oncogene. 2015; 34: 5175-86.
96.Francescone R, Hou V, Grivennikov SI. Microbiome, inflammation and cancer. Cancer J. 2014; 20: 181-9.
97.Vendramini-Costa DB, Francescone R, Posocco D, Hou V, Dmitrieva O, Hensley H, et al. Anti-inflammatory natural product goniothalamin reduces colitis-associated and sporadic colorectal tumorigenesis. Carcinogenesis. 2017; 38: 51-63.
98.Ma Y, Galluzzi L, Zitvogel L, Kroemer G. Autophagy and cellular immune responses. Immunity. 2013; 39: 211-27.
99.Braun T, Carvalho G, Fabre C, Grosjean J, Fenaux P, Kroemer G. Targeting NF-κB in hematologic malignancies. Cell Death Differ. 2006; 13: 748-58.
100.Cilloni D, Martinelli G, Messa F, Baccarani M, Saglio G. Nuclear factor κB as a target for new drug development in myeloid malignancies. Haematologica. 2007; 92: 1224-9.
101.Pérez L, Muñoz-Durango N, Riedel CA, Echeverría C, Kalergis AM, Cabello-Verrugio C, et al. Endothelial-to-mesenchymal transition: Cytokine-mediated pathways that determine endothelial fibrosis under inflammatory conditions. Cytokine Growth Factor Rev. 2017; 33: 41-54.
102.Copland JA, Sheffield-Moore M, Koldzic-Zivanovic N, Gentry S, Lamprou G, Tzortzatou-Stathopoulou F, et al. Sex steroid receptors in skeletal differentiation and epithelial neoplasia: is tissue-specific intervention possible? BioEssays. 2009; 31: 629-41.
103.Diefenbacher ME, Litfin M, Herrlich P, Kassel O. The nuclear isoform of the LIM domain protein Trip6 integrates activating and repressing signals at the promoter-bound glucocorticoid receptor. Mol Cell Endocrinol. 2010; 320: 58-66.
104.Chodankar R, Wu DY, Gerke DS, Stallcup MR. Selective coregulator function and restriction of steroid receptor chromatin occupancy by Hic-5. Mol Endocrinol. 2015; 29: 716-29.
105.Bonomo A, Monteiro AC, Gonçalves-Silva T, Cordeiro-Spinetti E, Galvani RG, Balduino A. A T cell view of the bone marrow. Front Immunol. 2016; 7: 184
106.Kortylewski M, Xin H, Kujawski M, Lee H, Liu Y, Harris T, et al. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell. 2009; 15: 114-23.
107.Kong HY, Zhang QW, Zeng YH, Wang H, Wu MQ, Zheng TY, et al. Prognostic significance of STAT3/phosphorylated-STAT3 in tumor: a meta-analysis of literatures. Int J Clin Exp Med. 2015; 8: 8525-39.
108.Lippitz BE, Harris RA. Cytokine patterns in cancer patients: A review of the correlation between interleukin 6 and prognosis. Oncoimmunology. 2016; 5: e1093722
109.Chen J, Li GS, Meng H, Fan YC, Song YH, Wang SR, et al. Upregulation of B7-H1 expression is associated with macrophage infiltration in hepatocellular carcinomas. Cancer Immunol. 2012; 61: 101-8.
110.Shostak K, Zhang X, Hubert P, Göktuna SI, Jiang ZS, Klevernic I, et al. NF-κB-induced KIAA1199 promotes survival through EGFRsignalling. Nat Commun. 2014; 5: 5232
111.Chou CH, Cheng YF, Siow TY, Kumar A, Peck K, Chang C. SCUBE3 regulation of early lung cancer angiogenesis and metastatic progression. Clin Exp Metastasis. 2013; 30: 741-52.
112.Lin KL, Cheng JN, Yang T, Li YS, Zhu B. EGFR-TKI downregulates PD-L1 in EGFR mutant NSCLC through inhibiting NF-κB. Biochem Biophys Res Commun. 2015; 463: 95-101.
113.Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS, et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci Transl Med. 2017; 9: eaal3604
114.Geginat J, Larghi P, Paroni M, Nizzoli G, Penatti A, Pagani M, et al. The light and the dark sides of Interleukin-10 in immunemediated diseases and cancer. Cytokine Growth Factor Rev. 2016; 30: 87-93.
115.Keating R, McGargill MA. mTOR regulation of lymphoid cells in immunity to pathogens. Front Immunol. 2016; 7: 180
116.Moschovi M, Adamaki M, Vlahopoulos SA. Progress in treatment of viral infections in children with acute lymphoblastic leukemia. Oncol Rev. 2016; 10: 300
117.Montecalvo A, Watkins SC, Orange J, Kane LP. Inducible turnover of optineurin regulates T cell activation. Mol Immunol. 2017; 85: 9-17.
118.Kuwahara I, Lillehoj EP, Koga T, Isohama Y, Miyata T, Kim KC. The signaling pathway involved in neutrophil elastase stimulated MUC1 transcription. Am J Respir Cell Mol Biol. 2007; 37: 691-8.
119.Jang JE, Eom JI, Jeung HK, Cheong JW, Lee JY, Kim JS, et al. Targeting AMPK-ULK1-mediated autophagy for combating BET inhibitor resistance in acute myeloid leukemia stem cells. Autophagy. 2017; 13: 761-2. doi: 10.1080/15548627.2016.1278328.
120.Chandrika G, Natesh K, Ranade D, Chugh A, Shastry P. Suppression of the invasive potential of Glioblastoma cells by mTOR inhibitors involves modulation of NFκB and PKC-α signaling. Sci Rep. 2016; 6: 22455
121.Grafone T, Palmisano M, Nicci C, Storti S. An overview on the role of FLT3-tyrosine kinase receptor in acute myeloid leukemia: biology and treatment. Oncol Rev. 2012; 6: e8
122.Gross SM, Rotwein P. Mapping growth-factor-modulated Akt signaling dynamics. J Cell Sci. 2016; 129: 2052-63.
123.Li S, Pinard M, Wang YL, Yang L, Lin RT, Hiscott J, et al. Crosstalk between the TNF and IGF pathways enhances NF-κB activation and signaling in cancer cells. Growth Horm IGF Res. 2015; 25: 253-61.
124.Baik IH, Jo GH, Seo D, Ko MJ, Cho CH, Lee MG, et al. Knockdown of RPL9 expression inhibits colorectal carcinoma growth via the inactivation of Id-1/NF-κB signaling axis. Int J Oncol. 2016; 49: 1953-62.
125.Gu X, Hua ZY, Dong YD, Zhan Y, Zhang XW, Tian W, et al. Proteome and acetylome analysis identifies novel pathways and targets regulated by perifosine in Neuroblastoma. Sci Rep. 2017; 7: 42062
126.Guan ZF, Li C, Fan JH, He DL, Li L. Androgen receptor (AR) signaling promotes RCC progression via increased endothelial cell proliferation and recruitment by modulating AKT→NF-κB→CXCL5 signaling. Sci Rep. 2016; 6: 37085
127.Lin YW, Lee LM, Lee WJ, Chu CY, Tan P, Yang YC, et al. Melatonin inhibits MMP-9 transactivation and renal cell carcinoma metastasis by suppressing Akt-MAPKs pathway and NF-κB DNA-binding activity. J Pineal Res. 2016; 60: 277-90.
128.Hu L, Shi Y, Hsu JH, Gera J, Van Ness B, Lichtenstein A. Downstream effectors of oncogenic ras in multiple myeloma cells. Blood. 2003; 101: 3126-35.
129.Fiskus W, Sharma S, Qi J, Shah B, Devaraj SGT, Leveque C, et al. BET protein antagonist JQ1 is synergistically lethal with FLT3 tyrosine kinase inhibitor (TKI) and overcomes resistance to FLT3-TKI in AML cells expressing FLT-ITD. Mol Cancer Ther. 2014; 13: 2315-27.
130.Zhu B, Zhang JB, Chen J, Li CL, Wang XD. Molecular biological characteristics of the recruitment of hematopoietic stem cells from bone marrow niche in chronic myeloid leukemia. Int J Clin Exp Pathol. 2015; 8: 12595-607.
131.Safaee M, Ivan ME, Oh MC, Oh T, Sayegh ET, Kaur G, et al. The role of epidermal growth factor-like module containing mucinlike hormone receptor 2 in human cancers. Oncol Rev. 2014; 8: 242
132.Bresalier RS, Ho SB, Schoeppner HL, Kim YS, Sleisenger MH, Brodt P, et al. Enhanced sialylation of mucin-associated carbohydrate structures in human colon cancer metastasis. Gastroenterology. 1996; 110: 1354-67.
133.Zhu GF, Huang Y, Wu CT, Wei D, Shi YX. Activation of G-protein-coupled estrogen receptor inhibits the migration of human nonsmall cell lung cancer cells via IKK-β/NF-κB signals. DNA Cell Biol. 2016; 35: 434-42.
134.Lee CJ, Lee MH, Yoo SM, Choi KI, Song JH, Jang JH, et al. Magnolin inhibits cell migration and invasion by targeting the ERKs/RSK2 signaling pathway. BMC Cancer. 2015; 15: 576
135.Guarneri C, Bevelacqua V, Polesel J, Falzone L, Cannavò PS, Spandidos DA, et al. NF κB inhibition is associated with OPN/MMP 9 downregulation in cutaneous melanoma. Oncol Rep. 2017; 37: 737-46.
136.Szebeni GJ, Vizler C, Kitajka K, Puskas LG. Inflammation and cancer: extra- and intracellular determinants of tumor-associated macrophages as tumor promoters. Mediators Inflamm. 2017; 2017: 9294018 doi: 10.1155/2017/9294018.
137.Salmiheimo A, Mustonen H, Vainionpää S, Shen Z, Kemppainen E, Puolakkainen P, et al. Tumour-associated macrophages activate migration and STAT3 in pancreatic ductal adenocarcinoma cells in co-cultures. Pancreatology. 2017; doi: 10.1016/j.pan.2017.04.013.
138.Huang J, Shen FR, Huang HT, Ling CH, Zhang GB. Th1high in tumor microenvironment is an indicator of poor prognosis for patients with NSCLC. Oncotarget. 2017; 8: 13116-25. doi: 10.18632/oncotarget.14471.
139.Meng J, Zhang XT, Liu XL, Fan L, Li C, Sun Y, et al. WSTF promotes proliferation and invasion of lung cancer cells by inducing EMT via PI3K/Akt and IL-6/STAT3 signaling pathways.Cell Signal. 2016; 28: 1673-82.
140.Abdelhamed S, Ogura K, Yokoyama S, Saiki I, Hayakawa Y. AKTSTAT3 pathway as a downstream target of EGFR signaling to regulate PD-L1 expression on NSCLC cells. J Cancer. 2016; 7: 1579-86.
141.Zhang N, Zeng YY, Du WW, Zhu JJ, Shen D, Liu ZY, et al. The EGFR pathway is involved in the regulation of PD-L1 expression via the IL-6/JAK/STAT3 signaling pathway in EGFR-mutated non-small cell lung cancer. Int J Oncol. 2016; 49: 1360-8.
142.Sumimoto H, Takano A, Teramoto K, Daigo Y. RAS-mitogenactivated protein kinase signal is required for enhanced PD-L1 expression in human lung cancers. PLoS One. 2016; 11: e0166626
143.Huang Q, Han JL, Fan JS, Duan LM, Guo MF, Lv ZL, et al. IL-17 induces EMT via Stat3 in lung adenocarcinoma. Am J Cancer Res. 2016; 6: 440-51.
144.Dong WL, Sun SW, Cao XC, Cui YH, Chen A, Li X, et al. Exposure to TNF-α combined with TGF-β induces carcinogenesis in vitro via NF-κB/Twist axis. Oncol Rep. 2017; 37: 1873-82.
145.Wamsley JJ, Kumar M, Allison DF, Clift SH, Holzknecht CM, Szymura SJ, et al. Activin upregulation by NF-κB is required to maintain mesenchymal features of cancer stem-like cells in nonsmall cell lung cancer. Cancer Res. 2015; 75: 426-35.
146.Keglowich L, Roth M, Philippova M, Resink T, Tjin G, Oliver B, et al. Bronchial smooth muscle cells of asthmatics promote angiogenesis through elevated secretion of CXC-chemokines (ENA-78, GRO-α, and IL-8). PLoS One. 2013; 8: e81494
147.Phillips KLE, Cullen K, Chiverton N, Michael ALR, Cole AA, Breakwell LM, et al. Potential roles of cytokines and chemokines in human intervertebral disc degeneration: interleukin-1 is a master regulator of catabolic processes. Osteoarthritis Cartilage. 2015; 23: 1165-77.
148.Singha B, Gatla HR, Manna S, Chang TP, Sanacora S, Poltoratsky V, et al. Proteasome inhibition increases recruitment of IκB kinase β (IKKβ), S536P-p65, and transcription factor EGR1 to interleukin-8 (IL-8) promoter, resulting in increased IL-8 production in ovarian cancer cells. J Biol Chem. 2014; 289: 2687-700.
149.Zhang L, Shao LG, Creighton CJ, Zhang YQ, Xin L, Ittmann M, et al. Function of phosphorylation of NF-kB p65 ser536 in prostate cancer oncogenesis. Oncotarget. 2015; 6: 6281-94.
150.Mizutani T, Ishizaka A, Furuichi Y. The werner protein acts as a coactivator of nuclear factor κB (NF-κB) on HIV-1 and interleukin-8 (IL-8) promoters. J Biol Chem. 2015; 290: 18391-9.
151.Alam M, Rajabi H, Ahmad R, Jin CN, Kufe D. Targeting the MUC1-C oncoprotein inhibits self-renewal capacity of breast cancer cells. Oncotarget. 2014; 5: 2622-34.
152.Jiang L, Yin M, Wei XX, Liu JX, Wang XH, Niu C, et al. Bach1 represses Wnt/β-catenin signaling and angiogenesis. Circ Res. 2015; 117: 364-75.
153.Jang J, Ha JH, Chung SI, Yoon Y. Β-catenin regulates NF-κB activity and inflammatory cytokine expression in bronchial epithelial cells treated with lipopolysaccharide. Int J Mol Med. 2014; 34: 632-8.
154.Kim KN, Ko SC, Ye BR, Kim MS, Kim J, Ko EY, et al. 5-Bromo-2-hydroxy-4-methyl-benzaldehyde inhibited LPS-induced production of pro-inflammatory mediators through the inactivation of ERK, p38, and NF-κB pathways in RAW 264.7 macrophages. Chem Biol Interact. 2016; 258: 108-14.
155.Rahman MM, Alkhouri H, Tang F, Che WC, Ge Q, Ammit AJ. Sphingosine 1-phosphate induces neutrophil chemoattractant IL-8: repression by steroids. PLoS One. 2014; 9: e92466
156.Hsieh YY, Shen CH, Huang WS, Chin CC, Kuo YH, Hsieh MC, et al. Resistin-induced stromal cell-derived factor-1 expression through Toll-like receptor 4 and activation of p38 MAPK/ NFκB signaling pathway in gastric cancer cells. J Biomed Sci. 2014; 21: 59
157.Clifford RL, Patel JK, John AE, Tatler AL, Mazengarb L, Brightling CE, et al. CXCL8 histone H3 acetylation is dysfunctional in airway smooth muscle in asthma: regulation by BET. Am J Physiol Lung Cell Mol Physiol. 2015; 308: L962-72.
158.Wadhwa E, Nicolaides T. Bromodomain inhibitor review: bromodomain and extra-terminal family protein inhibitors as a potential new therapy in central nervous system tumors. Cureus. 2016; 8: e620
159.Shi J, Whyte WA, Zepeda-Mendoza CJ, Milazzo JP, Shen C, Roe JS, et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 2013; 27: 2648-62.
160.Perry MM, Durham AL, Austin PJ, Adcock IM, Chung KF. BET bromodomains regulate transforming growth factor-β-induced proliferation and cytokine release in asthmatic airway smooth muscle. J Biol Chem. 2015; 290: 9111-21.
161.Sionov RV, Vlahopoulos SA, Granot Z. Regulation of Bim in health and disease. Oncotarget. 2015; 6: 23058-134.
162.Mehla K, Singh PK. MUC1: a novel metabolic master regulator. Biochim Biophys Acta. 2014; 1845: 126-35.
163.Bouillez A, Rajabi H, Pitroda S, Jin CN, Alam M, Kharbanda A, et al. Inhibition of MUC1-C suppresses MYC expression and attenuates malignant growth in KRAS mutant lung adenocarcinomas. Cancer Res. 2016; 76: 1538-48.
164.Tagde A, Rajabi H, Stroopinsky D, Gali R, Alam M, Bouillez A, et al. MUC1-C induces DNA methyltransferase 1 and represses tumor suppressor genes in acute myeloid leukemia. Oncotarget. 2016; 7: 38974-87.
165.Kato K, Uchino R, Lillehoj EP, Knox K, Lin Y, Kim KC. Membrane-tethered MUC1 mucin counter-regulates the phagocytic activity of macrophages. Am J Respir Cell Mol Biol. 2016; 54: 515-23.
166.Barham W, Chen LY, Tikhomirov O, Onishko H, Gleaves L, Stricker TP, et al. Aberrant activation of NF-κB signaling in mammary epithelium leads to abnormal growth and ductal carcinoma in situ. BMC Cancer. 2015; 15: 647
167.Liu X, Caffrey TC, Steele MM, Mohr A, Singh PK, Radhakrishnan P, et al. MUC1 regulates cyclin D1 gene expression through p120 catenin and β-catenin. Oncogenesis. 2014; 3: e107
168.Li Y, Yi HY, Yao YX, Liao XD, Xie YQ, Yang J, et al. Thecytoplasmic domain of MUC1 induces hyperplasia in the mammary gland and correlates with nuclear accumulation of βcatenin. PLoS One. 2011; 6: e19102
169.Kufe DW. MUC1-C oncoprotein as a target in breast cancer: activation of signaling pathways and therapeutic approaches. Oncogene. 2013; 32: 1073-81.
170.Nishida A, Lau CW, Zhang M, Andoh A, Shi HN, Mizoguchi E, et al. The membrane-bound mucin Muc1 regulates T helper 17-cell responses and colitis in mice. Gastroenterology. 2012; 142: 865-74.
171.Gaemers IC, Vos HL, Volders HH, van der Valk SW, Hilkens J. A stat-responsive element in the promoter of the episialin/MUC1 gene is involved in its overexpression in carcinoma cells. J Biol Chem. 2001; 276: 6191-9.
172.Banerjee S, Mujumdar N, Dudeja V, Mackenzie T, Krosch TK, Sangwan V, et al. MUC1c regulates cell survival in pancreatic cancer by preventing lysosomal permeabilization. PLoS One. 2012; 7: e43020
173.Vlahopoulos S, Critselis E, Voutsas IF, Perez SA, Moschovi M, Baxevanis CN, et al. New use for old drugs? Prospective targets of chloroquines in cancer therapy Curr Drug Targets. 2014; 15: 843-51.
174.Huang WC, Chan ML, Chen MJ, Tsai TH, Chen YJ. Modulation of macrophage polarization and lung cancer cell stemness by MUC1 and development of a related small-molecule inhibitor pterostilbene. Oncotarget. 2016; 7: 39363-75.
175.Wu S, Ge YL, Huang LQ, Liu HY, Xue Y, Zhao Y. BRG1, the ATPase subunit of SWI/SNF chromatin remodeling complex, interacts with HDAC2 to modulate telomerase expression in human cancer cells. Cell Cycle. 2014; 13: 2869-78.
176.Zhou Z, Su Y, Fa X. Restoration of BRG1 inhibits proliferation and metastasis of lung cancer by regulating tumor suppressor miR-148b. OncoTargets Ther. 2015; 8: 3603-12.
177.Obeid S, Hebbard L. Role of adiponectin and its receptors in cancer. Cancer Biol Med. 2012; 9: 213-20.
178.Wu D, Wu P, Zhao L, Huang L, Zhang Z, Zhao S, et al. NF-κB Expression and Outcomes in Solid Tumors: A Systematic Review and Meta-Analysis. Medicine. 2015; 94: e1687
179.Liao C, Yu ZB, Guo W, Liu QY, Wu YN, Li YF, et al. Prognostic value of circulating inflammatory factors in non-small cell lung cancer: a systematic review and meta-analysis. Cancer Biomark. 2014; 14: 469-81.
180.Xu YH, Lu S. A meta-analysis of STAT3 and phospho-STAT3 expression and survival of patients with non-small-cell lung cancer. Eur J Surg Oncol. 2014; 40: 311-7.
181.Marrugal Á, Ojeda L, Paz-Ares L, Molina-Pinelo S, Ferrer I. Proteomic-based approaches for the study of cytokines in lung cancer. Dis Markers. 2016; 2016: 2138627
182.Fang YY, Bi FF, Zhou YM, Sun WP, Li CY, Liu Q, et al. Nicotinamide adenine dinucleotide (NAD) may affect DNA methyltransferase 1 through regulation of BRCA1 in ovarian cancer. Am J Cancer Res. 2015; 5: 1199-206.
183.Buckley NE, Haddock P, De Matos Simoes R, Parkes E, Irwin G, Emmert-Streib F, et al. A BRCA1 deficient, NFκB driven immune signal predicts good outcome in triple negative breast cancer. Oncotarget. 2016; 7: 19884-96.
Cite this article as: Vlahopoulos SA. Aberrant control of NF-κB in cancer permits transcriptional and phenotypic plasticity, to curtail dependence on host tissue: molecular mode. Cancer Biol Med. 2017; 14: 254-70. doi: 10.20892/j.issn.2095-3941.2017.0029
Spiros A. Vlahopoulos
E-mail: v_spiros@hotmail.com
March 18, 2017; accepted June 8, 2017.
Available at www.cancerbiomed.org
Copyright © 2017 by Cancer Biology & Medicine
 Cancer Biology & Medicine2017年3期
Cancer Biology & Medicine2017年3期
- Cancer Biology & Medicine的其它文章
- Survival after pulmonary metastasectomy in colorectal cancer patients: does a history of resected liver metastases worsen the prognosis? A literature review
- Expression levels of β-catenin and galectin-3 in meningioma and their effect on brain invasion and recurrence: a tissue microarray study
- Potential predictive factors for pathologic complete response after the neoadjuvant treatment of rectal adenocarcinoma: a single center experience
- Inhibition of IKK-NFκB pathway sensitizes lung cancer cell lines to radiation
- BsmI (rs1544410) and FokI (rs2228570) vitamin D receptor polymorphisms, smoking, and body mass index as risk factors of cutaneous malignant melanoma in northeast Italy
- Preclinical and clinical applications of specific molecular imaging for HER2-positive breast cancer