
Nanotechnology-based combination therapy for overcoming multidrug-resistant cancer
2017-08-27 03:24:21MengZhangErgangLiuYannaCuiYongzhuoHuang
Cancer Biology & Medicine 2017年3期
Meng Zhang, Ergang Liu, Yanna Cui, Yongzhuo Huang
1Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China;2University of Chinese Academy of Sciences, Beijing 100049, China;3State Key Laboratory of Chemical Engineering, School of Chemical Engineering and Technology, Tianjin University, Tianjin 300072, China;4Key Laboratory of Primate Neurobiology, Institute of Neuroscience, Chinese Academy of Sciences, Shanghai 200031, China
REVIEW
Nanotechnology-based combination therapy for overcoming multidrug-resistant cancer
Meng Zhang1,2*, Ergang Liu1,3*, Yanna Cui1,4, Yongzhuo Huang1,2
1Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China;2University of Chinese Academy of Sciences, Beijing 100049, China;3State Key Laboratory of Chemical Engineering, School of Chemical Engineering and Technology, Tianjin University, Tianjin 300072, China;4Key Laboratory of Primate Neurobiology, Institute of Neuroscience, Chinese Academy of Sciences, Shanghai 200031, China
Multidrug resistance (MDR) is a major obstacle to successful cancer treatment and is crucial to cancer metastasis and relapse. Combination therapy is an effective strategy for overcoming MDR. However, the different pharmacokinetic (PK) profiles of combined drugs often undermine the combination effect in vivo, especially when greatly different physicochemical properties (e.g., those of macromolecules and small drugs) combine. To address this issue, nanotechnology-based codelivery techniques have been actively explored. They possess great advantages for tumor targeting, controlled drug release, and identical drug PK profiles. Thus, a powerful tool for combination therapy is provided, and the translation from in vitro to in vivo is facilitated. In this review, we present a summary of various combination strategies for overcoming MDR and the nanotechnology-based combination therapy.
Drug delivery; nanotechnology; multidrug resistance; combination therapy; cancer therapy
Introduction
Cancer is one of the leading causes of global death. According to the WHO, up to 8.2 million people had died from malignant tumors in 2012, accounting for 22% of all noncommunicable disease-related deaths1. As a fatal threat to health, the long-lasting battle between human and cancer can be dated back to 160 years ago, and the “weapons” had been evolving with time, i.e., from the original caustics2,3to various synthetic/natural cytotoxins4, later the inhibitors targeting specific intracellular pathways with less side effect5, and molecular tools that can silence the carcinogenic genes had been developed and applied in cancer treatment6. To date, the total number of antitumor drugs marketed can be counted in hundreds, which have benefited numerous patients. The medication can produce significant therapeutic responses and lead to remission in lipid cancers, such as acute myeloid leukemia and lymphoma, as well as solid tumors7,8. However, they might serve as a “selective stress” that can induce the proliferation of the therapy-resistant cancer cells and finally reshape the cancer geometry9,10. As a result, tumor sensitivity to therapeutics gradually decreases and multidrug resistance (MDR) eventually develops10. The emerged MDR plays a crucial role in tumor metastasis and relapse11, accounting for approximately 10%–90% of the clinical recurrences (varying among different types of cancer) during the following three years after the initial remission12. Of note, MDR accounts for over 90% of chemotherapy failures in patients with metastatic cancer13.
MDR can develop via different mechanisms, including increased drug efflux mediated by the overexpressed MDR-related transporters, increased DNA repair capacity, dysfunctional apoptosis, or activation of prosurvival pathways10,14(Figure 1). Furthermore, the genetic heterogeneity also represents an important factor, which contributed to tumor’s MDR against clinical medications4,15,16. Considering the complexity of MDR-inducing mechanisms, a combination of two or more drugs targeting different oncogenetic pathways is useful for overcoming MDR and improving the therapeutic index. Drug combination can be screened according to the mechanisms involved in MDR development. For example, chemotherapeutics may be combined with P-glycoprotein (P-gp) inhibitors, tyrosine kinase inhibitors (TKIs), orproapoptotic agents to enhance the cytotoxicity17.

Figure 1 Various mechanisms involved in tumor MDR. The major mechanisms include activated substituted signaling pathway, increased DNA repair capacity, dysfunctional apoptosis, drug efflux mediated by the MDR-related transporters, and decreased drug uptake.
A sharp increase of studies focusing on cancer combination therapy has been observed during the past 30 years, showing more than 35,000 relating works published during the recent 10 years in 2007–2016 (Figure 2A). Enormous efforts have been placed on screening the optimized combination formulations and exploring the related molecular mechanisms. Several aspects as follows must also be addressed in achieving an effective therapeutic response in vivo when applying the combination regimens screened from the in vitro tests.
(a) Pharmacokinetic (PK) diversity. Given that the PK behavior varies among different drugs, an effective dose ratio of the combined drugs, optimized by the cell-based tests, is hard to realize the optimal antitumor responses in vivo following systemic administration18.

Figure 2 Summary of articles published on “combination therapy of cancer” (A) and “nanotechnology-mediated combination therapy of cancer” (B). We summarized the retrieval number of the articles in the “Web of Science” database from 1947 to 2016 with the search terms“combination therapy and cancer” and 2007 to 2016 with the search terms “nano* and combination therapy and cancer”.
(b) Physiological variety. Drugs must undergo a series of PK processes before the arrival on their molecular targets for taking pharmacological action19. The physiological diversity in these processes (i.e., enzyme degradation, gastrointestinal absorption, serum protein binding, blood to tumor perfusion, and intracellular delivery) can lead to variation in the drug availability to the tumor cells.
(c) Dose regime. Sequential dosing is needed in certain circumstances when the chemical toxins are combined with a chemosensitizer, for instance. A sensitizer should be preadministrated to reverse the resistance of the tumor cells. The PK differences between the combined drugs, as well as dose timing, often make the treatments poorly predictable with combination regimentation20.
Nanotechnology-based codelivery
Nanotechnology-based drug delivery is a groundbreaking strategy that has changed the landscape of pharmacotherapy. The nanomaterials (NMs) have been extensively applied for PK improvement and site-specific delivery and thus provided a useful delivery tool for combination therapy21,22. First, by taking advantage of NMs, the encapsulated drugs can be simultaneously delivered. Thus, NMs help maintain a relatively identical in vivo fate and the dose ratio of the combined drugs. Second, NMs can preferentially accumulate at the tumor site via enhanced permeability and retention (EPR) effect and active targeting mechanisms. In addition, NMs can release the encapsulated drugs in a controlled manner. Thus, NMs are helpful in increasing tumor drug concentration and improving therapeutic efficacy. Third, NMs are resistant to drug efflux mediated by MDR transporters because of the size-exclusion effect, and thus the drugs can retain an effective intracellular concentration23. Fourth, the NMs can alter the drug distribution in organelles (e.g., nuclear targeting) and thereby increase the drug concentration in the targeting organelles and enhance the efficacy24. Fifth, some NMs also bear the bioactivity of inhibiting the proliferation of neoplasm cells (such as silver nanoparticles) and can display synergistic effect with the delivered drugs25.
In addition, the druggability can be improved by the nanotechnology. The development of cancer biology and drug discovery has identified thousands of therapeutics, many of which unfortunately failed to further develop into clinical drugs because of unfavorable druggability, such as poor solubility, low-membrane permeability, and instability in biological fluids. Meanwhile, the PK variations among different drugs would result in unwanted toxicity and variable therapeutic effects26,27. On this account, NMs have been demonstrated with the capacities of improving drug solubility and stability, as well as promoting the penetration through various biological barriers. Importantly, the NMs can help synchronize the delivery of the coencapsulated drugs and enhance the synergistic effect of the combined drugs.
Given the unique advantages of nanotechnology-based codelivery and its promising applications in anti-MDR cancer therapy, developing the combination strategies for overcoming MDR, such as the rational designs of NMs for optimal combined formulation with different drugs and the patterns of NM-mediated combination therapy, should be considered.
Targeting delivery
NMs can preferentially accumulate at the tumor site via EPR effect and active targeting mechanisms and subsequently release the encapsulated drugs in a controlled manner, thus providing the benefits of increasing the tumor drug concentration and the therapeutic efficacy. In general, the delivery efficiency of the encapsulated drugs to the target organs can be optimized by adjusting the physiochemical features of nanovehicles, such as shape, size, the surface hydrophilicity–hydrophobicity, and zeta potentials19. Further modification with targeting ligands is able to improve the delivery efficiency21. The targeting delivery is important for both promoting therapeutic effect and reducing adverse effect21,22,28. Specifically, NMs may preferentially enter the different subcellular compartments, and it is useful to deliver the specific drugs into certain organelles (e.g., nuclear targeting) and further enhance the therapeutic responses25.
Drug ratio maintenance
The optimal antitumor efficacy can be obtained in vitro by facilely optimizing the concentrations and molar ratio of the coadministrated drugs. However, its predictive in vivo application is difficult because of the various PK profiles that thereby result in the submaximal concentrations and a nonoptimum molar ratio of combined drugs. Notably, the altering molar ratio of combined drugs might even cause antagonistic effect, providing a challenge during combination therapy29. Nanotechnology facilitates the combination treatment by codelivery of the encapsulated drugs to the tumor, with a relatively identical in vivo fate and dose ratio of the combined drugs28. For example, the combination use of rinotecan and cisplatin displays an antagonistic regionbetween the molar ratios of 1:2 to 4:1, and beyond this ratio range, the two drugs yield synergistic effect30. By coencapsulating the two drugs into liposomes, the fixed drug ratio can be maintained for more than 24 h. However, for the liposomal formulations, several concerns of drug premature release in blood are observed, leading to unwanted effects. To precisely control the drug ratio and in vivo release behavior, the phosphorous lipids can be crosslinked by intermolecular disulfide bonds. By using this cross-linked multilamellar liposome vesicle (cMLV), the drug ratio of the coencapsulated doxorubicin (DOX) and paclitaxel (PTX) was precisely maintained for more than 24 h, maximizing the therapeutic effect while minimizing the systemic toxicity31.
The ratio control of sunitinib and curcumin can be achieved by using the bovine serum albumin (BSA)-coated superparamagnetic iron oxide nanoparticles (SPIOs) in a xenograft tumor-bearing mouse model32. In vitro formulation screening results showed that the optimal drug ratio of sunitinib to curcumin was approximately 0.5. Deviation from this ratio would lead to the increased combination index value and compromise the synergistic effect. By coencapsulating the two drugs in the protein layers, the sunitinib/curcumin ratio was maintained in an optimal range. Meanwhile, the nanoformulated drugs manifested a significantly increased tumor accumulation (29.8-fold for sunitinib and 8.4-fold for curcumin compared with free drugs), showing the dual benefits of SPIO-mediated combination therapy (Figure 3).
NM-mediated MDR inhibition
Aside from the functions of improving the PK behavior, some NMs can function as P-gp inhibitors and restore the antineoplastic activities of the loading drugs in the MDR cells. The NMs may reverse MDR effect via different ways. For example, inorganic silver nanoparticles are resistant to drug efflux because of the size-exclusion effect23. Many amphiphilic copolymers can inhibit the activities of overexpressed P-gp pumps. For example, Pluronic P123/F127, methoxy poly (ethylene glycol)–poly (lactide) copolymer (mPEG-PLA), and TPGS 1000 (D-R-tocopheryl polyethylene glycol 1000 succinate) can inhibit the P-gp by ATP depletion33-35. However, surface modification using TPGS does not induce ATP depletion-associated P-gp inhibition. Instead, the anti-MDR effect of TPGS-modified PLA nanoparticles can be attributed to the increased drug uptake and intracellular protection from enzyme degradation36,37. Liposomal drugs have been reported with effects on altering the raft compositions in the resistant cells and decreasing the lipid raft-associated P-gp and the DOX-loaded liposomes, thus impaired the transport and ATPase activity of P-gp38.
Despite the various mechanisms involved in P-gp inhibition, NMs may restore tumor’s sensitivity by simply enhancing the cellular uptake of the payload drugs. For example, Liu and co-workers used a cMLV for codelivery of DOX and PTX into the drug-resistant 4T1 cells39. The results showed that neither single drug-loaded cMLV nor soluble drug mixtures can inhibit P-gp expression, whereas the dual drug-loaded cMLV can efficiently suppress P-gp expression and cell proliferation. The enhanced uptake mediated by the cMLV yielded the sufficiently high intracellular concentration of both drugs and their coaction thus inhibited the P-gp function of 4T1 cells.
Tailing drug release to achieve the optimal synergistic effect
The synergy of combined drugs for certain instances requires the drugs to be given in a sequential manner. For example, sequential treatment with erlotinib and DOX can rewire the apoptotic signaling pathway and increase antineoplastic effects on the triple-negative BT-20 cells20. Similar trends can be found in a combination use of chemotherapeutics with RNAi agents. Given that the target depletion effect of RNAi generally occurs at 48 h after the RNAi treatment, the coloaded RNAi agent and antitumor drugs should be released sequentially40. Of note, sequential or simultaneous dosing regimentation depends on the specified synergistic patterns of the combined drugs. For example, sequential application of verapamil (P-gp inhibitor) and vincristine (antitumor agent) in treating MCF7/adverse drug reaction (ADR) cancer did not yield superior antitumor effect compared with the simultaneous administration41. However, the sequential application of verapamil and vincristine displayed higher efficacy than the reverse sequential application (i.e., vincristine→verapamil). As known, MDR can be effectively reversed by P-gp inhibitors, such as verapamil, tariquidar, zosuquidar, and laniquidar42, and combination of cytotoxins with MDR inhibitors may significantly increase intracellular drug retention and enhanced cytotoxicity43,44. The results indicated that small molecular P-gp inhibitors can lead to immediate inactivation of the drug efflux pump. Thus, sequential application of chemodrugs with P-gp inhibitors is not needed.

Figure 3 Ratio controlled delivery of sunitinib and curcumin by BSA-stabilized SPIOs. (A) Preparation scheme of the BSA-stabilized SPIOs and subsequent encapsulation of sunitinib and curcumin (SPIO-SC). (B) Sunitinib distribution in the major organs. (C) Distribution of curcumin in the major organs. (D) Combination index (CI) value of sunitinib and curcumin at different Sun/Cur ratios. CI > 1, antagonistic effect; CI = 1, additivity; CI < 1, synergistic effect. (E) Dynamic variation of the SunCur ratio in the tumor over time post administration. (F) Increased drug retention of SPIO-SC in the tumor compared with SunCur. Reprinted with permission from Ref. 32.
For combination therapy of the vascular disruptive combretastatin A4 (CA4) with chemodrugs, however, sequential release pattern is optimal45. The number of blood vessels decreased in 72 h after applying CA4. The subsequent use of antineoplasmic agents also enhanced the EPR effect of tumor and yielded an increased tumor uptake and improved therapeutic response. Inspired by this effect, several kinds of nanovehicles including liposome, poly (ethylene glycol)-block-poly (D,L-lactic acid) (PEG-b-PLA) micelle, and mesoporous silica nanoparticles (MSNs), have been developed for sequential delivery of CA4 and antineoplastic drugs. In the liposomal formulation, hydrophobic CA4 and hydrophilic DOX•HCl were separately encapsulated into thelipid bilayer and inner aqueous phase of RGD liposomes. The release of aqueous DOX in the inner phase was confined by the lipid bilayer because of the hydrophilic differences, and a differential release of DOX and CA4 from the liposome was achieved46. In the micellar formulation, DOX or PTX was covalently conjugated with the hydrophobic terminal of the copolymeric PEG-b-PLA. Hydrophobic CA4 was also incorporated into the micellar cores. On this account, the release kinetics of the payloads was dependent on the binding pattern between the micelles and the encapsulated drugs. The release of CA4 in the hydrophobic cores of micelles was relatively faster than the polymer-conjugated DOX or PTX47,48. Another interesting application was the coencapsulation in the MSNs. The hydrophilic CA4P (combretastatin phosphate) and DOX were encapsulated by MSNs via physical adsorption. CA4, due to the difference in surface charge, was quickly released from the negatively charged MSNs because of electrostatic repulsion, whereas positively charged DOX was released in a slow pattern49.
In addition, the benefit of simultaneous application of a chemodrug and a sensitizer was also demonstrated in treating cancer stem cells. Sun et al.50reported that simultaneous delivery of a differentiation agent all-trans-retinoic acid (ATRA) with a chemodrug DOX in the same nanovehicle can efficiently induce cancer stem cell (CSC) differentiation and tumor suppression. In general, CSC is a promising target for cancer therapy51. Being self-renewal and resistant to drug interference, CSC is considered as a major mechanism responsible for MDR. In this case, combining chemical drugs with the differential stimuli agents can drive CSC into differentiation and restore sensitivity to chemotherapy52. As reported, the key to avoiding CSC enrichment was simultaneously delivering ATRA and DOX using the same carriers50.
However, the principle of sequential release control during combination use of chemotherapeutics with their sensitizers is hard to achieve when multiple dosing is needed because the steady-state plasma drug concentration (plateau concentration) of both drugs after multiple administrations will attenuate the benefits from sequential regimentation53.
Methods to coencapsulate drugs with different physiochemical properties
Drugs can be encapsulated into NMs via different methods, such as adsorption, conjugation or encapsulation. Surface adsorption of drugs into the NMs is advantageous for the facile preparation. In addition, the drug release pattern is typically characterized by a rapid and burst release kinetics. In comparison, encapsulation inside the NMs may synchronize the drug release in response to NM degradation54. Thereby, the release rate is tunable by strategically selecting the materials with varying degradation rates. Furthermore, drugs can be covalently conjugated to the NMs via the cleavable bonds/linkers, by which the drug release is dependent on the cleavage of the bonds/linkers55-57.
Nanotechnology-based combination therapy
Combination of cytotoxins/cytotoxins
In general, the cytotoxic drugs can be categorized into four groups, namely, cell cycle-independent alkylating agent (e.g., cisplatin, tetrazine, and alkylate nucleoids), cell cycledependent antimetabolites (e.g., methotrexate, pemetrexed, and gemcitabine), cell-cycle dependent antimicrotubule agents (e.g., PTX and vincristine), and topoisomerase inhibitors (e.g., DOX and etoposide). Combination application of two or more chemical toxins with different pharmacological actions may synergistically decrease cell viability and is the most widely used strategy in clinical practice. Chemical drugs are diverse in solubility, hydrophobicity, and PK behavior, providing difficulty to maintain the in vivo molecular ratio of the combined drugs in the tumor. Furthermore, most small-molecular-weight chemotherapeutics are the substrates of the MDR transporters58, which can significantly attenuate their antitumor effect. Overall, the nanovehicles should be carefully designed to take advantage of the merits of all combined drugs and avoid unfavorable metabolisms.
A large amount of functional NMs have been developed, bearing various properties in structure, physicochemical characteristics, as well as the loading pattern of drug payloads, enabling the modulation of release behavior of the encapsulated drugs.
(a) Stimulus-responsive NMs. Applications of “smart”NMs can facilitate the encapsulated drugs to be released in a stimulus-sensitive manner. For example, the MSNs are advantageous for drug loading but suffer from nonspecific leakage and burst release of the encapsulated drugs. To overcome this problem, the pH-responsive polymers, such as methacrylic-type ionic liquid terpolymer59, or thermosensitive poly (N-isopropylacrylamide) (PNIPAM)60can be grafted into the surface of the MSNs and serve as the switchable “gate keepers”, enabling the encapsulated drugs to be released in response to the tumor microenvironment (TME). For example, a polyelectrolyte multilayer-modified MSN was constructed via coating poly (allylaminehydrochloride) and poly (styrene sulfonate) into the surface of MSNs layer by layer with the DOX encapsulated into the pore of the MSNs under pH 261. This system can respond to low pH and release the DOX at pH 5.0 but without drug release at pH 7.4. Chen et al.62modified the azide into the SiO2surface and synthesized the PNIPAM via the reversible addition-fragmentation transfer polymerization of N-isopropylacrylamide monomer. Then, a thermos-responsive nanoparticle system was constructed by grafting PNIPAM to the SiO2surface based on the azide-alkyne cycloaddition.
(b) Hybrid NMs. Coadministration of two NMs loading with different drugs (one in each) has been explored for achieving synchronic delivery. However, even encapsulated by the same kind of NMs, the optimum synergistic effect may not be guaranteed if the two drugs are separately loaded47. This challenge can be addressed by using the hybrid NMs–the different drug loading nanoparticles can be crosslinked together or using the core–shell-structured nanoparticles. Depending on the structure of the hybrid NMs, the payloads may be simultaneously released from the hybrid NMs or be sequentially released from a core–shellstructured NM63. For example, for sequential delivery of DOX and PTX, the DOX-loaded mesoporous nanoparticles were first prepared, then the surface was coated with polylactic-co-glycolic acid as PTX-loading layer64.
Combination of cytotoxins and molecularly targeted agents
The mutations in the cancer cell signal pathways provide the pathological basis for targeted therapies by applying the inhibitors to selectively block the mutated signal pathways that are crucial in tumorigenesis. Therefore, the molecularly targeted agents display the improved therapeutic efficacy but reduced systemic toxicity compared with the chemotherapeutics. However, cancer cells also develop drug resistance to these molecularly targeted agents via the mechanisms including upregulation of the therapeutic target, activating the alternative compensatory survival signaling pathways, and inactivating the cell death signaling pathways65. Novel combination strategies should be designed to disturb these mechanisms to reverse the resistance to the targeted drugs.
For example, the combination of cytotoxins and epidermal growth factor receptor (EGFR)-TKIs has been commonly used. The EGFR–tyrosine kinase pathway is a clinical therapeutic target for several types of cancers66. The pathway can be blocked by antiEGFR antibodies (e.g., cetuximab), or by small molecular drugs (e.g., erlotinib and sunitinib) to inhibit the intracellular-mutated tyrosine kinase. The combination with cytotoxins may synergistically inhibit the proliferation of tumor cells. Furthermore, certain TKIs may help rewire the apoptotic pathways and sensitize cells to cytotoxins67, or inhibit P-gp activity of tumor cells, thus increasing the efficacy of cytotoxic drugs68.
(a) Combination of cytotoxins/TKIs. Nanotechnology can help increase the tumor drug accumulation and overcome acquired drug resistance. For example, lapatinib, an inhibitor of EGFR and HER-2, has been approved by the FDA to treat HER-2 positive refractory breast cancers combined with various therapeutic agents, such as capecitabine, anthracyclines, taxanes, and trastuzumab. Lapatinib has also been reported with the ability to inhibit the activity of drug efflux transporters. Based on the amphiphilic poly (ethylene glycol)-block-poly (2-methyl-2-benzoxycarbonylpropylene carbonate) polymers, we developed the DOX micelles and lapatinib micelles, simultaneously, to codeliver DOX and lapatinib in treating multidrug-resistant breast cancer69. The results of fluorescence microscopy and flow cytometry showed that coadministration of the DOX micelles and lapatinib micelles can enhance DOX uptake in the MCF-7/ADR cells overexpressing drug efflux transporters but not MCF-7 cells low-expressing drug efflux transporters. Coinciding with the uptake result, codelivery of DOX micelles and lapatinib micelles significantly improved the in vitro antitumor efficacy in the MCF-7/ADR cells. For in vivo treatment, lapatinib can restore the sensitivity of breast tumor to DOX and reduce the systemic toxicity of DOX (Figure 4).
(b) Combination of cytotoxin/antivasculature agents. Combination of cytotoxic drugs with antivasculature drugs may be synergistically effective. Zhang et al.46,47have developed two independent systems (RGD-liposomes and PEG-b-PLA mixed micelles) for codelivery of the cytotoxic DOX with the antivasculature agent combretastatin A4 (CA4). In both systems, CA4 was observed to be released from the NMs much faster than DOX, which can destroy the vascular walls and facilitate the extraversion of post released DOX, thus increasing the therapeutic DOX efficacy.
Combination of cytotoxins and sensitizing agents

Figure 4 Codelivery DOX and lapatinib (LAPA) by polymer-based micelles. (A) Scheme of codelivery DOX and LAPA by polymer-based micelles in treating resistant breast tumor. (B) Improved therapeutic efficacy of coadministration of DOX micelles and LAPA micelles in xenograft MCF-7/ADR tumor-bearing mice model, and (C) the tumor images. Reprinted with permission from Ref. 69.
(a) Combination of cytotoxins with MDR inhibitors. MDR inhibitors, such as verapamil, tariquidar, zosuquidar, and laniquidar42, are commonly used as chemosensitizers for blocking the drug efflux transporters and thus restoring the sensitivity of tumor cells to chemotherapeutics. The combination of cytotoxins with MDR inhibitors may significantly increase intracellular drug retention and improve tumor killing effect43,44. NMs can improve the poor selectivity and low affinity of these inhibitors and achieve satisfactory synergistic results57,70. Qin et al.71encapsulated DOX and verapamil into the hydrogel nanoparticles. Coadministration of DOX-NPs and verapamil-NPs can significantly improve the uptake and DOX cytotoxicity in NCI/ADR-RES cells.
To sensitize tumor cells to the cytotoxic drugs, the cells/tumor need to be pretreated with the sensitizing agents. To achieve this process, NMs with sequential release behavior should be designed and utilized for the combined delivery of toxins and sensitizing drugs67. However, pretreatment may not be a requisite for all sensitizing agents.
(b) Combination of cytotoxins with immune regulators. TME plays an important role in cancer development, MDR, and metastasis. Therefore, remodeling TME is a potential target for overcoming MDR. We developed a mannosylated albumin nanoparticle system for codelivery of the cytotoxic agents disulfiram/copper complex and the M2 macrophage modulator regorafenib72. Given that the albumin-bindingprotein (e.g., SPARC) pathway and mannose receptor (MR) were highly expressed in both the drug-resistant colon tumor cells and M2 macrophages, such system can achieve dual targeting to both cell-membrane receptors and both cells. The “one-stone-two-bird” delivery strategy can significantly enhance the delivery efficiency and treatment efficacy against the drug-resistant colon cancer both in vitro and in vivo.
Combination of cytotoxins and peptides (or proteins)
With the advances of biotechnology, proteins and peptides have been widely investigated in cancer therapy because of their high specificity and efficacy73-75.
(a) Cytotoxin/therapeutic antibodies. Therapeutic antibodies (e.g., cetuximab, rituximab, and trastuzumab) represent an important class of protein/peptide drugs capable of inhibition cell proliferation by selectively binding to their membrane receptors76. The combination with antiserum or trastuzumab (HER2 antibody) markedly benefit the therapeutic effect of cytotoxins77,78. Of note, antibody-drug conjugates have shown significant therapeutic effects in clinical cancer treatment and attracted worldwide attention. This area has been specially reviewed by several articles79,80.
(b) Cytotoxin/apoptotic peptide. Certain intrinsic or extrinsic peptide/protein toxins are capable of inducing apoptosis of cancer cells, which may synergize cytotoxicity of chemotherapeutics. For example, N7 peptide of second mitochondria- derived activator (Smac N7) can bind with inhibitor of apoptosis and activate proapoptotic pathway81-83. When combined with cytotoxic PTX, N7 peptide may effectively promote PTX-induced toxicity, indicating a synergistic effect existed between Smac N7 and PTX84.
By rational design, chimeric peptides with unique physiochemical properties can serve as a potent therapeutic agent and as novel functional NMs for drug loading. The apoptotic peptide AVPI was a small hydrophobic peptide with poor membrane permeability. To address this problem, we designed a novel chimeric peptide containing AVPI and a cell penetrating peptide R8, wherein the added R8 peptide not only improved the water solubility of AVPI and its membrane permeability but also simultaneously acted as the DNA binding site85,86. The chimeric peptide and p53 DNA would self-assemble into nanoparticles through the electrostatic interaction between R8 and DNA, thus forming a codelivery system of the therapeutic AVPI and p53 DNA. The prepared cell-penetrating AVPIR8/p53 DNA nanocomplex can significantly increase the sensitivity of the resistant MCF-7/ADR cells to DOX. The in vivo therapeutic results showed that coadministration of the AVPIR8/p53 DNA nanocomplex with additional mixing with DOX can effectively inhibit the tumor growth with a reduced DOX dose and produce less side effect (Figure 5). Similarly, Li et al.82designed a novel amphiphilic peptide derivative with its hydrophilic part composed of Smac N7 peptide and a cell penetrating peptide and hydrophobic part being composed of four aliphatic tails. The peptide derivative can selfassemble into micelles and encapsulate DOX for combination therapy.
(c) Cytotoxins/protein toxins. Some protein toxins (e.g., recombinant trichosanthin) have been reported with the ability to kill the MDR cancer cells, as well as reverse the resistance of MDR cancer cells to chemotherapeutics. However, immunogenicity, enzymatic degradation, and poor membrane permeability are challenges for the application of such protein drugs87. Unlike small molecular drugs, macromolecular protein drugs that actually are in a nanosized scale can passively accumulate in the tumor site via EPR effect88. PEGylation and ligand modification further extend the blood circulation and tumor targeting89. However, PEGylation can reduce the membrane permeability of proteins. We previously developed a PEGylated, matrix metallopeptidase 2 (MMP2)-activatable cell-penetrating protein toxin trichosanthin (TCS) (termed rTLM-PEG) based on a recombinant intein-mediated site-specific conjugation method90. The protein system can dePEGylate in the TME via the enzymatic activation of matrix metalloproteinase to the substrate peptide and thus release the cell penetrating TCS. Such protein delivery system was further investigated for overcoming the drug-resistant lung cancer in combination with the PTX liposomes91. TCS effectively restored the sensitivity of A549/T cancer cells to PTX (Figure 6B). The mechanisms involved the inhibition of the caspase 9 phosphorylation and promotion of the caspase 3-dependent apoptosis (Figure 6C). TCS coadministration with PTX liposomes entirely arrested the tumor growth on A549/T tumor-bearing mouse model (Figure 6D).
Combination of chemotherapeutics and nucleic acid drugs
Tumorigenesis involves multiple genetic mutations92. The therapeutic stress drives further mutations and aggravates the changes of tumor geometry, which is partially accounted for acquired resistance to cancer therapy93. Various molecular tools, such as small interfering RNA, short hairpin RNA, and plasmid DNA, enable the precise regulation of the expression of a specific gene because of the development of molecularbiotechnology. However, the short half-life in blood, enzymatic degradation, renal clearance, and poor membrane permeability are major obstacles for clinical applications of nucleic acid drugs. Furthermore, single gene therapy may notbe sufficient to achieve the satisfactory treatment outcomes, leading to MDR94. Thus, combination therapy with two or more gene cocktails (e.g., targeting EGFR, MDR-1, bcl-2, and survivin) and cytotoxins can further improve the therapeutic efficacy95, and the nanotechnology would facilitate the application.
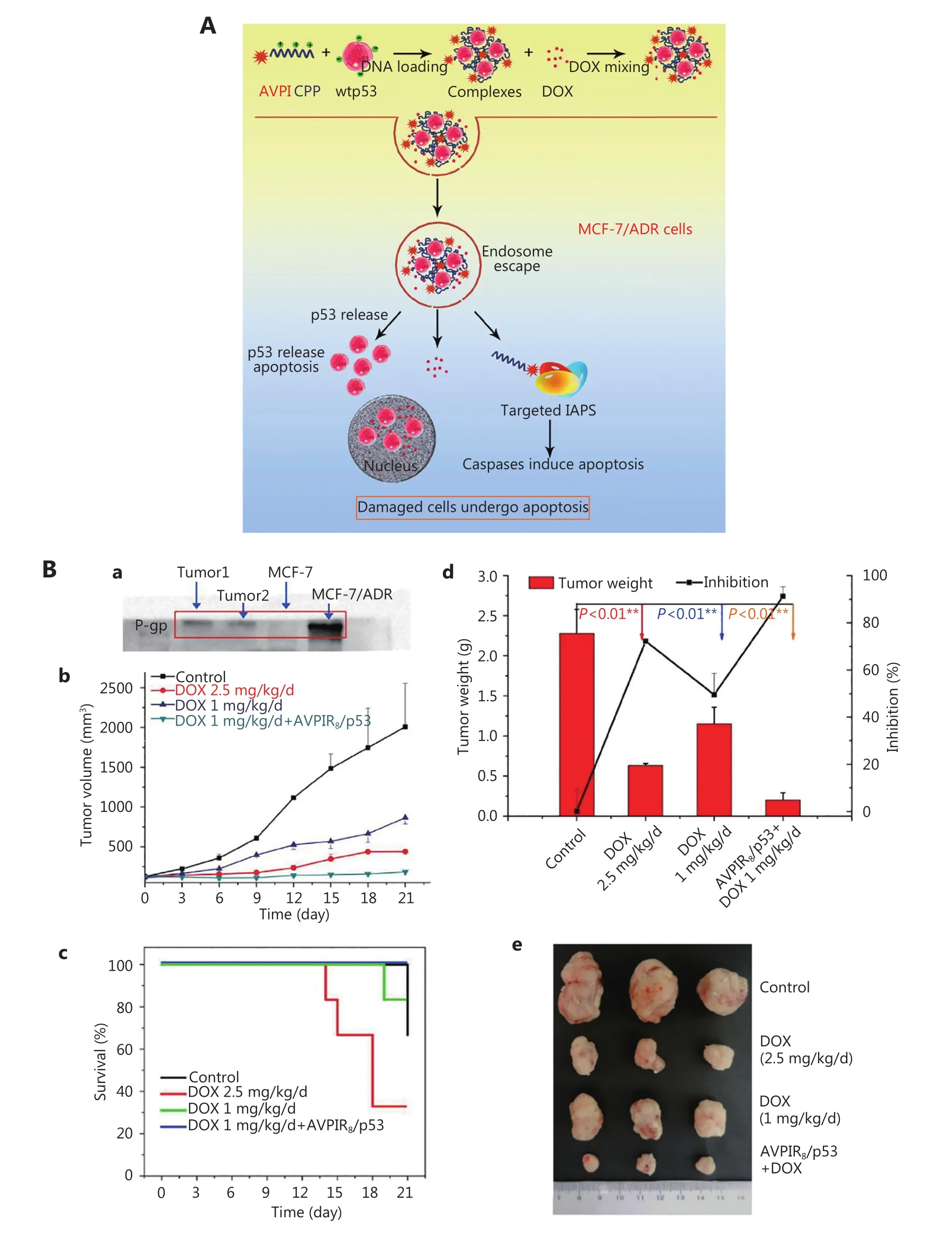
Figure 5 Cell-penetrating AVPIR8/p53 DNA nanocomplex as adjuvant therapy of cytotoxic agent DOX to overcome drug-resistant breast cancer. (A) Scheme of the cell-penetrating AVPIR8/p53 DNA nanocomplex combined with DOX to overcome MDR. (B) Improved therapy effect and reduced side effect of the cell-penetrating AVPIR8/p53 DNA nanocomplex with DOX. Reprinted with permission from Ref. 86.

Figure 6 Combination of PEGylated, MMP2-activatable cell-penetrating TCS with PTX liposomes for overcoming the drug-resistant lung cancer. (A) Scheme of the PEGylated, MMP2-activatable cell-penetrating TCS combined with PTX for overcoming drug resistance in A549/T cells. (B) In vitro synergistic cytotoxicity of PEGylated, MMP2-activatable cell-penetrating TCS, and PTX. (C) Regulatory effect of PEGylated, MMP2-activatable cell-penetrating TCS and PTX on caspase 9 phosphorylation and caspase 3. (D) Synergistic therapeutic effect of the PEGylated, MMP2-activatable cell-penetrating TCS and PTX liposomes in vivo. Reprinted with permission from Ref. 91.
(a) Cytotoxins and p53 gene combination. p53 is the“gatekeeper of the genome” and can regulate cell apoptosis through transcription-dependent and transcriptionindependent pathways by activating the expression of proapoptosis proteins and suppressing the activity of antiapoptosis proteins. Dysfunction of TP53 tumor suppressor is a main mechanism that cancer cells escape apoptosis and become insensitive to drugs96. Delivery of wild-type p53 is a promising strategy to restore cancer cells sensitive to therapeutic agents. p53 DNA combined with DOX86,97and PTX can significantly improve the therapeutic effect98. Adenovirus-mediated p53 gene (Ad-p53) transfection can restore the sensitivity of MCF-7/ADR cells to DOX99.
(b) Cytotoxins and Bcl-2 siRNA. RNA interference technology has offered a flexible tool for selective silencing tumorigenic genes. siRNA codelivery that knocks down the drug efflux transporters and antiapoptosis genes can restore cancer cells sensitive to chemotherapeutic drugs100. By codelivery of DOX with the siRNA targeting the Bcl-2 gene using the folate-targeted nanocarrier, DOX-induced apoptosis in the SKOV-3 cells overexpressing folate receptor was significantly enhanced through a mechanism of downregulating the antiapoptotic protein Bcl-2. In parallel,the proapoptotic protein Bax was also upregulated101.
(c) Cytotoxins and MDR siRNA. siRNA coadministration targeting the P-gp mRNA can improve the sensitivity of cancer cells to the chemotherapeutic agents. The codelivery system of antiP-gp siRNA and DOX displayed a synergistic effect in DOX-resistant cell line HepG2/adriamycin and xenograft tumor model102. P-gp downregulation can resensitize the DOX-resistance cancer cells to DOX103. Nanoparticle-mediated P-gp targeted siRNA and chemodrug PTX can effectively silence the MDR-1 gene and obviously increase the PTX accumulation104. Other MDR proteins, such as the major vault protein, can serve as a target with siRNA to downregulate the expression and yield the increased efficacy of the conventional cytotoxins105.
(d) Cytotoxins and survivin siRNA. Survivin plays an important role in cancer carcinogenesis and angiogenesis106. Silencing survivin with small hairpin RNA was found to be in concert with PTX treatment107. Torchilin and coworkers demonstrated that the survivin siRNA can sensitize the tumor cells to PTX108.
Other combinations
Other combinations (e.g., chemotherapy/thermotherapy, chemotherapy/phototherapy, and chemotherapy/radiotherapy) based on the nanotechnology have also been actively investigated. For example, nanographene oxide (NGO) and gold nanostructures can be used as cancer drug carriers and for photothermal ablation of tumor based on their photothermal transition properties. The NGO nanocomposites to deliver DOX showed the high therapeutic efficacy both in vitro and in vivo via the combination of the photothermal therapy of the NGOs and chemotherapy of DOX109,110.
Immunotherapy is a promising method in treating cancer, in which dendritic cells (DCs) are the major target. Cancer cells can suppress the maturation and function of DCs, which often results in the tumor immune tolerance. Therefore, immunotherapy is an attractive strategy to promote maturing DCs using immune-stimulatory factors. Chemotherapeutics (e.g., PTX) displayed a “Yin-and-Yang” nature that highdose chemotherapeutics compromised the functions of immune cells, whereas the low dose promoted and stimulated the DC maturation, thus exhibiting the bidirectional modulation of suppression and activation. We have recently reported the polymeric nanoassembly system for microneedle-assisted codelivery of pTRP-2 vaccine targeting the epidermis DCs and the immunomodulatory low-dose PTX for enhanced cancer immunotherapy111.
Cytosine-guanosine (CpG) oligodeoxynucleotides is a common immunostimulatory factor in clinical practice for melanoma. Yu Tao and coworkers designed the gold nanorods–CpG–DOX conjugates to achieve combined cancer therapy, including immunotherapy, photothermal therapy, and chemotherapy112. Gold nanoparticles can also enhance the tumor radiosensitivity during radiotherapy because of its high absorption capability to X-ray113-115.
Another interesting application is the combination of the bioactive functional NMs and drugs. Silver nanoparticles (AgNPs) possess potent antitumor activity and thus are used as therapeutic agents23,25. Ostad and coworkers revealed the combination effect of AgNPs and tamoxifen, wherein AgNPs combined with tamoxifen can effectively kill the parent and tamoxifen-resistant cells and reduce the tamoxifen doses116.
Prospects
MDR is a crucial challenge in antitumor therapy. As one of the most important strategies to address this problem, combination therapy has achieved remarkable progress in clinical cancer treatment. However, the conventional combination has been suffering from the varying PKs of different drugs, leading to inconsistent therapeutic responses during in vitro to in vivo translation. On this account, NMs with their potentials in drug coencapsulation, targeting delivery, controlled release, and PK improvement provide powerful tools for drug combination therapy.
However, several issues need to be addressed in the future development. First, although the nanocarriers can synchronize the PK behavior of the combined drugs, drug ratio in the tumor may also change because of the complexity of in vivo process57,117. Second, despite that NMs may preferentially accumulate in the tumors, the overall tumor retention is still limited, accounting for merely 1%–5% of the administered dose118. Thus, improving the efficacy of the NM-based combination therapy is greatly required. Third, most of NMs are highly entrapped by the RES organs (e.g., the liver, lung, and spleen), imposing potential safety issues. For example, to inhibit the P-gp-mediated drug resistance, cytotoxins were coencapsulated with MDR inhibitors, which can facilitate the drug uptake in the tumors but also increase the risk of hepatotoxicity. Fourth, long-term exposure of combined drugs may also lead to resistance119. Therefore, the NMs for combination therapy need to be multivalent and switchable, and drug replacement should be achieved readily. Fifth, nucleic acid drugs represent the effective means to overcome drug resistance in cancer therapy. However, the transport efficiency of nonvirus vectors is still limited. Sixth,the large-scale production of nanomedicine with a precise ratio control is a challenge. A marketed nanomedicine product with coencapsulation of drugs is still not available. In a word, nanotechnology has provided the convenient tools for combination therapy. However, nanotechnology still needs much improvement for clinical translation.
Acknowledgements
This work was supported by the grants from the National Basic Research Program of China (Grant No. 973 Program 2014CB931900, 2013CB932503) and National Natural Science Foundation of China (Grant No. 81373357, 81422048, 81673382, 81521005).
Conflict of interest statement
No potential conflicts of interest are disclosed.
1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015; 136: E359-86.
2.Fell JW. A treatise on cancer, and its treatment. London: J. Churchill, 1857.
3.Walshe WH. The nature and treatment of cancer. London: Taylor and Walton, 1846.
4.Kunjachan S, Rychlik B, Storm G, Kiessling F, Lammers T. Multidrug resistance: physiological principles and nanomedical solutions. Adv Drug Deliv Rev. 2013; 65: 1852-65.
5.Tsuruo T, Naito M, Tomida A, Fujita N, Mashima T, Sakamoto H, et al. Molecular targeting therapy of cancer: drug resistance, apoptosis and survival signal. Cancer Sci. 2003; 94: 15-21.
6.Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov. 2006; 5: 37-50.
7.Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016; 66: 271-89.
8.Zahreddine H, Borden KLB. Mechanisms and insights into drug resistance in cancer. Front Pharmacol. 2013; 4: 28
9.Labi V, Erlacher M. How cell death shapes cancer. Cell Death Dis. 2015; 6: e1675
10.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013; 13: 714-26.
11.Wu Q, Yang ZP, Nie YZ, Shi YQ, Fan D. Multi-drug resistance in cancer chemotherapeutics: mechanisms and lab approaches. Cancer Lett. 2014; 347: 159-66.
12.DeSantis CE, Lin CC, Mariotto AB, Siegel RL, Stein KD, Kramer JL, et al. Cancer treatment and survivorship statistics, 2014. CA Cancer J Clin. 2014; 64: 252-71.
13.Longley DB, Johnston PG. Molecular mechanisms of drug resistance. J Pathol. 2005; 205: 275-92.
14.Pan ST, Li ZL, He ZX, Qiu JX, Zhou SF. Molecular mechanisms for tumour resistance to chemotherapy. Clin Exp Pharmacol Physiol. 2016; 43: 723-37.
15.Kibria G, Hatakeyama H, Harashima H. Cancer multidrug resistance: mechanisms involved and strategies for circumvention using a drug delivery system. Arch Pharm Res. 2014; 37: 4-15.
16.Rebucci M, Michiels C. Molecular aspects of cancer cell resistance to chemotherapy. Biochem Pharmacol. 2013; 85: 1219-26.
17.Yap TA, Omlin A, de Bono JS. Development of therapeutic combinations targeting major cancer signaling pathways. J Clin Oncol. 2013; 31: 1592-605.
18.Cao P, Bae Y. Polymer nanoparticulate drug delivery and combination cancer therapy. Future Oncol. 2012; 8: 1471-80.
19.Rempe R, Cramer S, Qiao RR, Galla HJ. Strategies to overcome the barrier: use of nanoparticles as carriers and modulators of barrier properties. Cell Tissue Res. 2014; 355: 717-26.
20.Lee MJ, Ye AS, Gardino AK, Heijink AM, Sorger PK, MacBeath G, et al. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell. 2012; 149: 780-94.
21.Gao ZB, Zhang LN, Sun YJ. Nanotechnology applied to overcome tumor drug resistance. J Control Release. 2012; 162: 45-55.
22.Steichen SD, Caldorera-Moore M, Peppas NA. A review of current nanoparticle and targeting moieties for the delivery of cancer therapeutics. Eur J Pharm Sci. 2013; 48: 416-27.
23.Liu JH, Zhao YX, Guo QQ, Wang Z, Wang HY, Yang YX, et al. TAT-modified nanosilver for combating multidrug-resistant cancer. Biomaterials. 2012; 33: 6155-61.
24.Wang HX, Zhao YX, Wang HY, Gong JB, He HN, Shin MC, et al. Low-molecular-weight protamine-modified PLGA nanoparticles for overcoming drug-resistant breast cancer. J Control Release. 2014; 192: 47-56.
25.Liang JM, Zeng F, Zhang M, Pan ZZ, Chen YZ, Zeng YE, et al. Green synthesis of hyaluronic acid-based silver nanoparticles and their enhanced delivery to CD44+cancer cells. RSC Adv. 2015; 5: 43733-40.
26.Daniel DB, Ramachandran G, Swaminathan S. The challenges of pharmacokinetic variability of first-line anti-TB drugs. Expert Rev Clin Pharmacol. 2017; 10: 47-58.
27.Reynolds J, Heysell SK. Understanding pharmacokinetics to improve tuberculosis treatment outcome. Expert Opin Drug Metab Toxicol. 2014; 10: 813-23.
28.Ma L, Kohli M, Smith A. Nanoparticles for combination drug therapy. ACS Nano. 2013; 7: 9518-25.
29.Mayer LD, Janoff AS. Optimizing combination chemotherapy by controlling drug ratios. Mol Interv. 2007; 7: 216-23.
30.Tardi PG, Dos Santos N, Harasym TO, Johnstone SA, Zisman N, Tsang AW, et al. Drug ratio-dependent antitumor activity of irinotecan and cisplatin combinations in vitro and in vivo. Mol Cancer Ther. 2009; 8: 2266-75.
31.Liu YR, Fang JX, Kim YJ, Wong MK, Wang P. Codelivery ofdoxorubicin and paclitaxel by cross-linked multilamellar liposome enables synergistic antitumor activity. Mol Pharm. 2014; 11: 1651-61.
32.Chen SH, Liang QL, Liu EG, Yu ZL, Sun L, Ye JX, et al. Curcumin/sunitinib co-loaded BSA-stabilized SPIOs for synergistic combination therapy for breast cancer. J Mater Chem B. 2017; 5: 4060-72.
33.Wei Z, Yuan S, Hao JG, Fang XL. Mechanism of inhibition of P-glycoprotein mediated efflux by Pluronic P123/F127 block copolymers: relationship between copolymer concentration and inhibitory activity. Eur J Pharm Biopharm. 2013; 83: 266-74.
34.Li WJ, Li XR, Gao YJ, Zhou YX, Ma SJ, Zhao Y, et al. Inhibition mechanism of P-glycoprotein mediated efflux by mPEG-PLA and influence of PLA chain length on P-glycoprotein inhibition activity. Mol Pharm. 2014; 11: 71-80.
35.Collnot EM, Baldes C, Schaefer UF, Edgar KJ, Wempe MF, Lehr CM. Vitamin E TPGS P-glycoprotein inhibition mechanism: influence on conformational flexibility, intracellular ATP levels, and role of time and site of access. Mol Pharm. 2010; 7: 642-51.
36.Tan GR, Feng SS, Leong DT. The reduction of anti-cancer drug antagonism by the spatial protection of drugs with PLA-TPGS nanoparticles. Biomaterials. 2014; 35: 3044-51.
37.Li PY, Lai PS, Hung WC, Syu WJ. Poly(L-lactide)-vitamin E TPGS nanoparticles enhanced the cytotoxicity of doxorubicin in drugresistant MCF-7 breast cancer cells. Biomacromolecules. 2010; 11: 2576-82.
38.Riganti C, Voena C, Kopecka J, Corsetto PA, Montorfano G, Enrico E, et al. Liposome-encapsulated doxorubicin reverses drug resistance by inhibiting P-glycoprotein in human cancer cells. Mol Pharm. 2011; 8: 683-700.
39.Liu YR, Fang JX, Joo KI, Wong MK, Wang P. Codelivery of chemotherapeutics via crosslinked multilamellar liposomal vesicles to overcome multidrug resistance in tumor. PLoS One. 2014; 9: e110611
40.Tsouris V, Joo MK, Kim SH, Kwon IC, Won YY. Nano carriers that enable co-delivery of chemotherapy and RNAi agents for treatment of drug-resistant cancers. Biotechnol Adv. 2014; 32: 1037-50.
41.Song XR, Cai Z, Zheng Y, He G, Cui FY, Gong DQ, et al. Reversion of multidrug resistance by co-encapsulation of vincristine and verapamil in PLGA nanoparticles. Eur J Pharm Sci. 2009; 37: 300-5.
42.Thomas H, Coley HM. Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting pglycoprotein. Cancer Control. 2003; 10: 159-65.
43.Wang FH, Zhang DR, Zhang Q, Chen YX, Zheng DD, Hao LL, et al. Synergistic effect of folate-mediated targeting and verapamilmediated P-gp inhibition with paclitaxel -polymer micelles to overcome multi-drug resistance. Biomaterials. 2011; 32: 9444-56.
44.Patil Y, Sadhukha T, Ma LN, Panyam J. Nanoparticle-mediated simultaneous and targeted delivery of paclitaxel and tariquidar overcomes tumor drug resistance. J Control Release. 2009; 136: 21-9.
45.Mitrus I, Sochanik A, Cichoń T, Szala S. Combination of combretastatin A4 phosphate and doxorubicin-containing liposomes affects growth of B16-F10 tumors. Acta Biochim Pol. 2009; 56: 161-5.
46.Zhang YF, Wang JC, Bian DY, Zhang X, Zhang Q. Targeted delivery of RGD-modified liposomes encapsulating both combretastatin A-4 and doxorubicin for tumor therapy: in vitro and in vivo studies. Eur J Pharm Biopharm. 2010; 74: 467-73.
47.Yang TY, Wang YG, Li ZQ, Dai WB, Yin J, Liang L, et al. Targeted delivery of a combination therapy consisting of combretastatin A4 and low-dose doxorubicin against tumor neovasculature. Nanomedicine. 2012; 8: 81-92.
48.Wang Z, Ho PC. A nanocapsular combinatorial sequential drug delivery system for antiangiogenesis and anticancer activities. Biomaterials. 2010; 31: 7115-23.
49.Li XY, Wu MY, Pan LM, Shi JL. Tumor vascular-targeted codelivery of anti-angiogenesis and chemotherapeutic agents by mesoporous silica nanoparticle-based drug delivery system for synergetic therapy of tumor. Int J Nanomedicine. 2016; 11: 93-105.
50.Sun R, Liu Y, Li SY, Shen S, Du XJ, Xu CF, et al. Co-delivery of all-trans-retinoic acid and doxorubicin for cancer therapy with synergistic inhibition of cancer stem cells. Biomaterials. 2015; 37: 405-14.
51.Maccalli C, De Maria R. Cancer stem cells: perspectives for therapeutic targeting. Cancer Immunol Immunother. 2015; 64: 91-7.
52.Borst P. Cancer drug pan-resistance: pumps, cancer stem cells, quiescence, epithelial to mesenchymal transition, blocked cell death pathways, persisters or what? Open Biol. 2012; 2: 120066
53.Van Rossum JM. Pharmacokinetics of accumulation. J Pharm Sci. 1968; 57: 2162-5.
54.Wang Y, Liu EG, Sun XY, Huang PY, Long H, Wang H, et al. Pluronic L61 as a long-circulating modifier for enhanced liposomal delivery of cancer drugs. Polym Chem. 2013; 4: 2958-62.
55.N VR, Dinda VR, Ganivada MN, Das Sarma J, Shunmugam R. Efficient approach to prepare multiple chemotherapeutic agent conjugated nanocarrier. Chem Commun. 2014; 50: 13540-3.
56.Wang YP, Jiang YF, Zhang M, Tan J, Liang JM, Wang HX, et al. Protease-activatable hybrid nanoprobe for tumor imaging. Adv Funct Mater. 2014; 24: 5443-53.
57.Lammers T, Subr V, Ulbrich K, Peschke P, Huber PE, Hennink WE, et al. Simultaneous delivery of doxorubicin and gemcitabine to tumors in vivo using prototypic polymeric drug carriers. Biomaterials. 2009; 30: 3466-75.
58.Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006; 5: 219-34.
59.Salehi R, Hamishehkar H, Eskandani M, Mahkam M, Davaran S. Development of dual responsive nanocomposite for simultaneous delivery of anticancer drugs. J Drug Target. 2014; 22: 327-42.
60.You YZ, Kalebaila KK, Brock SL, Oupicky D. Temperaturecontrolled uptake and release in PNIPAM-modified porous silica nanoparticles. Chem Mater. 2008; 20: 3354-9.
61.Feng W, Zhou XJ, He CL, Qiu KX, Nie W, Chen L, et al. Polyelectrolyte multilayer functionalized mesoporous silica nanoparticles for pH-responsive drug delivery: layer thicknessdependent release profiles and biocompatibility. J Mater Chem B. 2013; 1: 5886-98.
62.Chen JC, Liu MZ, Chen C, Gong HH, Gao CM. Synthesis and characterization of silica nanoparticles with well-defined thermoresponsive PNIPAM via a combination of RAFT and click chemistry. ACS Appl Mater Interfaces. 2011; 3: 3215-23.
63.Su TT, Long YY, Deng CY, Feng LL, Zhang XL, Chen ZB, et al. Construction of a two-in-one liposomal system (TWOLips) for tumor-targeted combination therapy. Int J Pharm. 2014; 476: 241-52.
64.Cui YN, Xu QX, Chow PKH, Wang DP, Wang CH. Transferrinconjugated magnetic silica PLGA nanoparticles loaded with doxorubicin and paclitaxel for brain glioma treatment. Biomaterials. 2013; 34: 8511-20.
65.Baas T. Finding the perfect combination. SciBX. 2012; 5: doi:10.1038/scibx.2012.832.
66.Jackman AL, Kaye S, Workman P. The combination of cytotoxic and molecularly targeted therapies–can it be done? Drug Discov Today. 2004; 1: 445-54.
67.Morton SW, Lee MJ, Deng ZJ, Dreaden EC, Siouve E, Shopsowitz KE, et al. A nanoparticle-based combination chemotherapy delivery system for enhanced tumor killing by dynamic rewiring of signaling pathways. Sci Signal. 2014; 7: ra44
68.He M, Wei MJ. Reversing multidrug resistance by tyrosine kinase inhibitors. Chin J Cancer. 2012; 31: 126-33.
69.Wang HY, Li F, Du CG, Wang HX, Mahato RI, Huang YZ. Doxorubicin and lapatinib combination nanomedicine for treating resistant breast cancer. Mol Pharm. 2014; 11: 2600-11.
70.Wang JC, Goh B, Lu WL, Zhang Q, Chang A, Liu XY, et al. In vitro cytotoxicity of Stealth liposomes co-encapsulating doxorubicin and verapamil on doxorubicin-resistant tumor cells. Biol Pharm Bull. 2005; 28: 822-8.
71.Qin M, Lee YEK, Ray A, Kopelman R. Overcoming cancer multidrug resistance by codelivery of doxorubicin and verapamil with hydrogel nanoparticles. Macromol Biosci. 2014; 14: 1106-15.
72.Zhao PF, Yin WM, Wu AH, Tang YS, Wang JY, Pan ZZ, et al. Dual-targeting to cancer cells and M2 macrophages via biomimetic delivery of mannosylated albumin nanoparticles for drug-resistant cancer therapy. Adv Funct Mater. 2017. in Press
73.Patel A, Patel M, Yang XY, Mitra AK. Recent advances in protein and Peptide drug delivery: a special emphasis on polymeric nanoparticles. Protein Pept Lett. 2014; 21: 1102-20.
74.Witting M, Obst K, Friess W, Hedtrich S. Recent advances in topical delivery of proteins and peptides mediated by soft matter nanocarriers. Biotechnol Adv. 2015; 33: 1355-69.
75.Ye ML, Kim S, Park K. Issues in long-term protein delivery using biodegradable microparticles. J Control Release. 2010; 146: 241-60.
76.Adams GP, Weiner LM. Monoclonal antibody therapy of cancer. Nat Biotechnol. 2005; 23: 1147-57.
77.He Q, Gao H, Gao M, Qi SM, Zhang YQ, Wang JZ. Anti-gastrins antiserum combined with lowered dosage cytotoxic drugs to inhibit the growth of human gastric cancer SGC7901 cells in nude mice. J Cancer. 2015; 6: 448-56.
78.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001; 344: 783-92.
79.Lambert JM, Morris CQ. Antibody-drug conjugates (ADCs) for personalized treatment of solid tumors: a review. Adv Ther. 2017; 34: 1015-35.
80.Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017; 16: 315-37.
81.Wang SM. Design of small-molecule Smac mimetics as IAP antagonists//Vassilev L, Fry D. Small-molecule inhibitors of protein-protein interactions. Current Topics in Microbiology and Immunology, vol 348. Berlin: Springer, 2011: 89-113.
82.Li MX, Liu P, Gao GH, Deng JZ, Pan ZY, Wu X, et al. Smac therapeutic peptide nanoparticles inducing apoptosis of cancer cells for combination chemotherapy with doxorubicin. ACS Appl Mater Interfaces. 2015; 7: 8005-12.
83.Sun HY, Stuckey JA, Nikolovska-Coleska Z, Qin DG, Meagher JL, Qiu S, et al. Structure-based design, synthesis, evaluation, and crystallographic studies of conformationally constrained Smac mimetics as inhibitors of the X-linked inhibitor of apoptosis protein (XIAP). J Med Chem. 2008; 51: 7169-80.
84.Mao HL, Pang YX, Zhang XL, Yang F, Zheng JF, Wang Y, et al. Smac peptide potentiates TRAIL- or paclitaxel-mediated ovarian cancer cell death in vitro and in vivo. Oncol Rep. 2013; 29: 515-22.
85.Wang HY, Guo QQ, Jiang YF, Liu EG, Zhao YX, Wang HX, et al. Co-delivery of cell-permeable chimeric apoptosis AVPIR8peptide/p53 DNA for cocktail therapy. Adv Funct Mater. 2013; 23: 6068-75.
86.Wang HY, Wang HX, Liang JM, Jiang YF, Guo QQ, Peng HG, et al. Cell-penetrating apoptotic peptide/p53 DNA nanocomplex as adjuvant therapy for drug-resistant breast cancer. Mol Pharm. 2014; 11: 3352-60.
87.Ezan E. Pharmacokinetic studies of protein drugs: past, present and future. Adv Drug Deliv Rev. 2013; 65: 1065-73.
88.Torchilin V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv Drug Deliv Rev. 2011; 63: 131-5.
89.Roberts M, Bentley MD, Harris JM. Chemistry for peptide and protein PEGylation. Adv Drug Deliv Rev. 2012; 64: 116-27.
90.Chen YZ, Zhang M, Jin HY, Tang YS, Wang HY, Xu Q, et al. Intein-mediated site-specific synthesis of tumor-targeting protein delivery system: turning PEG dilemma into prodrug-like feature. Biomaterials. 2017; 116: 57-68.
91.Chen YZ, Zhang M, Jin HY, Tang YS, Wu AH, Xu Q, et al. Prodrug-like, PEGylated protein toxin trichosanthin for reversal of chemoresistance. Mol Pharm. 2017; 14: 1429-38.
92.Dancey JE, Bedard PL, Onetto N, Hudson TJ. The genetic basis for cancer treatment decisions. Cell. 2012; 148: 409-20.
93.Alexander S, Friedl P. Cancer invasion and resistance: interconnected processes of disease progression and therapyfailure. Trends Mol Med. 2012; 18: 13-26.
94.Pecot CV, Calin GA, Coleman RL, Lopez-Berestein G, Sood AK. RNA interference in the clinic: challenges and future directions. Nat Rev Cancer. 2011; 11: 59-67.
95.Yin Q, Shen JA, Chen LL, Zhang ZW, Gu WW, Li YP. Overcoming multidrug resistance by co-delivery of Mdr-1 and survivin-targeting RNA with reduction-responsible cationic poly(β-amino esters). Biomaterials. 2012; 33: 6495-506.
96.Bai L, Zhu WG. p53: structure, function and therapeutic applications. J Cancer Mol. 2006; 2: 141-53.
97.Wiradharma N, Tong YW, Yang YY. Self-assembled oligopeptide nanostructures for co-delivery of drug and gene with synergistic therapeutic effect. Biomaterials. 2009; 30: 3100-9.
98.Zhao F, Yin H, Li J. Supramolecular self-assembly forming a multifunctional synergistic system for targeted co-delivery of gene and drug. Biomaterials. 2014; 35: 1050-62.
99.Qi XD, Chang ZK, Song J, Gao G, Shen Z. Adenovirus-mediated p53 gene therapy reverses resistance of breast cancer cells to adriamycin. Anticancer Drugs. 2011; 22: 556-62.
100.Chen AM, Zhang M, Wei DG, Stueber D, Taratula O, Minko T, et al. Co-delivery of doxorubicin and Bcl-2 siRNA by mesoporous silica nanoparticles enhances the efficacy of chemotherapy in multidrug-resistant cancer cells. Small. 2009; 5: 2673-7.
101.Zou SY, Cao N, Cheng D, Zheng RQ, Wang J, Zhu KS, et al. Enhanced apoptosis of ovarian cancer cells via nanocarriermediated codelivery of siRNA and doxorubicin. Int J Nanomedicine. 2012; 7: 3823-35.
102.Zhang CG, Zhu WJ, Liu Y, Yuan ZQ, Yang SD, Chen WL, et al. Novel polymer micelle mediated co-delivery of doxorubicin and P-glycoprotein siRNA for reversal of multidrug resistance and synergistic tumor therapy. Sci Rep. 2016; 6: 23859
103.Xiong XB, Lavasanifar A. Traceable multifunctional micellar nanocarriers for cancer-targeted co-delivery of MDR-1 siRNA and doxorubicin. ACS Nano. 2011; 5: 5202-13.
104.Patil YB, Swaminathan SK, Sadhukha T, Ma LA, Panyam J. The use of nanoparticle-mediated targeted gene silencing and drug delivery to overcome tumor drug resistance. Biomaterials. 2010; 31: 358-65.
105.Han M, Lv Q, Tang XJ, Hu YL, Xu DH, Li FZ, et al. Overcoming drug resistance of MCF-7/ADR cells by altering intracellular distribution of doxorubicin via MVP knockdown with a novel siRNA polyamidoamine-hyaluronic acid complex. J Control Release. 2012; 163: 136-44.
106.Tu SP, Jiang XH, Lin MC, Cui JT, Yang Y, Lum CT, et al. Suppression of survivin expression inhibits in vivo tumorigenicity and angiogenesis in gastric cancer. Cancer Res. 2003; 63: 7724-32.
107.Hu QL, Li W, Hu XR, Hu QD, Shen J, Jin X, et al. Synergistic treatment of ovarian cancer by co-delivery of survivin shRNA and paclitaxel via supramolecular micellar assembly. Biomaterials. 2012; 33: 6580-91.
108.Salzano G, Riehle R, Navarro G, Perche F, De Rosa G, Torchilin VP. Polymeric micelles containing reversibly phospholipidmodified anti-survivin siRNA: a promising strategy to overcome drug resistance in cancer. Cancer Lett. 2014; 343: 224-31.
109.Qin XC, Guo ZY, Liu ZM, Zhang W, Wan MM, Yang BW. Folic acid-conjugated graphene oxide for cancer targeted chemophotothermal therapy. J Photochem Photobiol B. 2013; 120: 156-62.
110.Bai J, Liu Y, Jiang XE. Multifunctional PEG-GO/CuS nanocomposites for near-infrared chemo-photothermal therapy. Biomaterials. 2014; 35: 5805-13.
111.Xu JJ, Xu BH, Tao J, Yang YX, Hu Y, Huang YZ. Microneedleassisted, DC-targeted codelivery of pTRP-2 and adjuvant of paclitaxel for transcutaneous immunotherapy. Small. 2017, doi: 10.1002/smll.201700666. in press
112.Tao Y, Ju EG, Liu Z, Dong K, Ren JS, Qu XG. Engineered, selfassembled near-infrared photothermal agents for combined tumor immunotherapy and chemo-photothermal therapy. Biomaterials. 2014; 35: 6646-56.
113.Hainfeld JF, Slatkin DN, Smilowitz HM. The use of gold nanoparticles to enhance radiotherapy in mice. Phys Med Biol. 2004; 49: N309-15.
114.Mesbahi A. A review on gold nanoparticles radiosensitization effect in radiation therapy of cancer. Rep Pract Oncol Radiother. 2010; 15: 176-80.
115.Park J, Park J, Ju EJ, Park SS, Choi J, Lee JH, et al. Multifunctional hollow gold nanoparticles designed for triple combination therapy and CT imaging. J Control Release. 2015; 207: 77-85.
116.Ostad SN, Dehnad S, Nazari ZE, Fini ST, Mokhtari N, Shakibaie M, et al. Cytotoxic activities of silver nanoparticles and silver ions in parent and tamoxifen-resistant T47D human breast cancer cells and their combination effects with tamoxifen against resistant cells. Avicenna J Med Biotechnol. 2010; 2: 187-96.
117.Milane L, Duan ZF, Amiji M. Pharmacokinetics and biodistribution of lonidamine/paclitaxel loaded, EGFR-targeted nanoparticles in an orthotopic animal model of multi-drug resistant breast cancer. Nanomedicine. 2011; 7: 435-44.
118.Lee BK, Yun YH, Park K. Smart nanoparticles for drug delivery: boundaries and opportunities. Chem Eng Sci. 2015; 125: 158-64.
119.Villanueva MT. Therapeutic resistance: paradox breaking. Nat Rev Cancer. 2015; 15: 71
Cite this article as: Zhang M, Liu E, Cui Y, Huang Y. Nanotechnology-based combination therapy for overcoming multidrug-resistant cancer. Cancer Biol Med. 2017; 14: 212-27. doi: 10.20892/j.issn.2095-3941.2017.0054
*These authors have contributed equally to this work.
Yongzhuo Huang
E-mail: yzhuang@simm.ac.cn
May 4, 2017; accepted July 3, 2017.
Available at www.cancerbiomed.org
Copyright © 2017 by Cancer Biology & Medicine
 Cancer Biology & Medicine2017年3期
Cancer Biology & Medicine2017年3期
- Cancer Biology & Medicine的其它文章
- Survival after pulmonary metastasectomy in colorectal cancer patients: does a history of resected liver metastases worsen the prognosis? A literature review
- Expression levels of β-catenin and galectin-3 in meningioma and their effect on brain invasion and recurrence: a tissue microarray study
- Potential predictive factors for pathologic complete response after the neoadjuvant treatment of rectal adenocarcinoma: a single center experience
- Inhibition of IKK-NFκB pathway sensitizes lung cancer cell lines to radiation
- BsmI (rs1544410) and FokI (rs2228570) vitamin D receptor polymorphisms, smoking, and body mass index as risk factors of cutaneous malignant melanoma in northeast Italy
- Preclinical and clinical applications of specific molecular imaging for HER2-positive breast cancer