
2009至2016年中国甲型H1N1流感病毒神经氨酸酶基因进化分析及流行趋势预测
2017-08-17 10:04宋玥颜文娟方坤李小杉李伟卫平民
临床检验杂志 2017年7期
宋玥,颜文娟,方坤,李小杉,李伟,卫平民
(1.东南大学公共卫生学院,南京 210009;2.南京市疾病预防控制中心急性传染病防制与学校卫生科,南京 210009)
·调查研究·
2009至2016年中国甲型H1N1流感病毒神经氨酸酶基因进化分析及流行趋势预测
宋玥1,颜文娟1,方坤1,李小杉1,李伟2,卫平民1
(1.东南大学公共卫生学院,南京 210009;2.南京市疾病预防控制中心急性传染病防制与学校卫生科,南京 210009)
目的 分析2009至2016年中国甲型H1N1流感病毒神经氨酸酶基因(NA)进化特征,预测其流行趋势,为流感疫苗评价提供依据。方法 从全球共享禽流感数据倡议组织(GISAID)数据库及美国国家生物技术中心(NCBI)数据库下载甲型H1N1流感病毒NA基因序列1 141条。利用邻接法进行系统进化分析和遗传变异分析,利用Bayesian skyline Plot预测流行趋势。结果 2009至2016年中国甲型H1N1毒株NA基因与参考毒株的同源性逐年降低。遗传进化分析显示同一年份的毒株在系统进化树上基本呈现集中分布。除2012年,其余各年份的毒株均通过不同模型得到正向压力选择位点。动力学分析显示2017年甲型H1N1可能达到一个流行高峰。结论 甲型H1N1流感病毒NA基因所编码的氨基酸逐年变异,需进一步加强流感监测。
甲型H1N1流感;神经氨酸酶;分子特征;进化分析
甲型H1N1流感病毒[influenza A viruses(H1N1 09pdm)]是由禽流感、猪流感和人流感病毒3者基因重配而成[1-2],自感染暴发以来,H1N1(09pdm)在人群中一直引起较高的发病率,也因此受到国内外研究的广泛关注。
流感病毒基因组由8个片段构成,其中神经氨酸酶(neuraminidase,NA)是流感病毒在传播和扩散中最关键的酶。NA附着在流感病毒颗粒表面[3],具有水解唾液酸的活性,从而促使病毒的释放和扩散[4],且NA抑制剂在合成抗流感病毒药物中占主要地位。NA的抗原位点、糖基化位点和耐药位点等关键的氨基酸突变是病毒抗原性发生变异的重要基础[5-6],其基因的改变对流感疫情的发生与流行有着密切的影响。本研究通过分析2009年后甲型H1N1流感病毒在中国地区的流行态势,对NA核苷酸序列进行系统进化及氨基酸位点变异分析,为我国甲型H1N1流感的疾病防控及预测提供理论依据。
1 材料与方法
1.1 材料 收集2009年至2016年国家流感中心(http://www.cnic.org.cn/chn/)流感周报中国流感流行的数据,从美国国家生物技术中心流感病毒(National Center for Biotechnology Information,NCBI)数据库(http://www.ncbi.nlm.nih.gov/genomes/FLU/FLU.html)及全球共享禽流感数据倡议组织(The Global Initiative on Sharing All Influenza Data,GISAID)数据库(http://platform.gisaid.org/epi3/frontend)中下载中国地区甲型H1N1流感病毒NA基因序列,共1 141条。用Molecular Evolutionary Genetics Analysis (MEGA) version 6.0软件进行序列比对,并用邻接法绘制进化树,根据树中终末分支同源性较高的分支情况,删除部分同地区同年份的序列。删除重复的序列和部分同年份同地区同源性较高的序列,得到354条来自安徽、北京、重庆、福建、甘肃、广东、广西、贵州、海南、河北、黑龙江、河南、香港、湖北、湖南、江苏、吉林、辽宁、内蒙古、宁夏、青海、山西、陕西、山东、上海、四川、天津、西安、新疆、云南、浙江各省份的序列,包括2009年87条、2010年55条、2011年26条、2012年4条、2013年34条、2014年24条、2015年54条和2016年70条。
1.2 方法 采用MEGA version 6.0软件进行序列比对、氨基酸位点的变异分析,采用邻接法构建甲型H1N1病毒NA基因进化树(Bootstrap检验重复值设为1 000)。通过Data monkey在线软件的Single likelihood ancestor counting(SLAC)、Internal fixed effects likelihood(IFEL)和Random effects method(REL)模型进行选择性压力分析。利用Figtree v1.4.2(http://beast.bio.ed.ac.uk)读取系统进化树,以bootstrap值≥0.9判定为流行枝(Lineage)。采用BEAST v1.8.0软件的Bayesian skyline Plot(BSP)法构建Bayesian skyline reconstruction,进行动力学分析,模型为GTR+Relaxed clock(uncorrelated)+Bayesian skyline。MCMC分析链长为200 000 000,迭代频次为1 000,收敛评价标准为有效样本数(effective sampling size, ESS)>200。
2 结果
2.1 流行病学分布特征 从全国流感监测网络实验室监测数据显示,2009年至2016年全国甲型H1N1流感共发生4次流行高峰,分别是2009年5月至2010年2月、2011年1至3月、2012年12月至2014年3月、2015年末至2016年又呈现上升趋势。见图1。
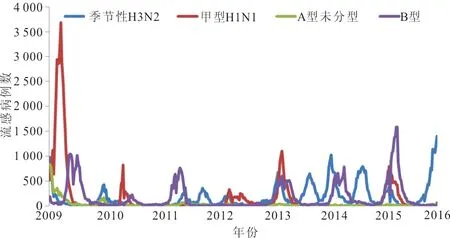
图1 中国地区2009至2016年流感流行情况
2.2 同源性分析 将不同年份甲型H1N1流感毒株(354条)与参考毒株A/California/07/2009(H1N1)的NA基因序列进行比对,其中2009至2016年的NA基因与参考毒株的同源性分别为99.2%~99.7%、99.2%~99.6%、98.8%~99.5%、98.8%~98.9%、98.4%~99.6%、98.2%~98.9%、97.9%~99.6%和97.5%~98.9%。2009至2016年甲型H1N1流感毒株与参考毒株的NA基因核苷酸和氨基酸遗传距离及各年内核苷酸的平均遗传距离见表1。
2.3 系统进化分析 甲型H1N1亚型NA基因系统发育进化树,如图2所示(图中的进化树只包含部分毒株,剔除了部分位于进化树上相同终末分支并且相同年份、同源性较高的序列,其中标为红色分支的是参考序列),最终选取了有代表性的71支毒株建树,不同年份的毒株由不同的颜色表示。结果显示,同一年份的甲型H1N1流感毒株在系统进化树上基本呈集中分布的态势。其中2009年与2010年的流感毒株形成一个分支;2011年的流感毒株另外形成2个小分支,可能是由于V241I和N369K的变异;2012年毒株N200S突变,独立形成一个小分支;2013年至2016年的毒株聚集形成一个大支。毒株间存在明显的年代差异,随着年代相隔越久,序列差异越大。
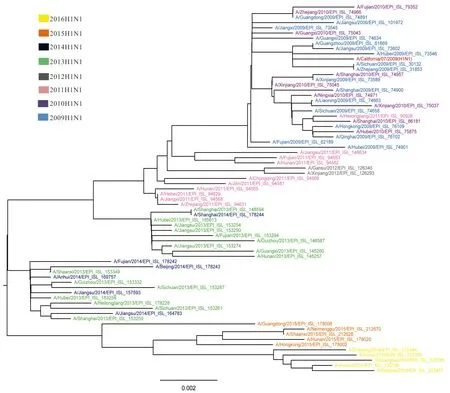
注:标尺值0.002代表毒株间的进化距离。
图2 中国2009-2016年甲型H1N1流感毒株NA基因系统进化树
2.4 NA分子特征分析
2.4.1 潜在抗原位点的氨基酸序列分析 NA蛋白N端第140至157位氨基酸很有可能是甲型H1N1流感病毒抗原决定簇所在[4]。中国地区的354条毒株序列与疫苗株A/California/07/2009(H1N1)相比,2010年潜在抗原位点氨基酸发生变异(G147E);2015年潜在抗原位点发生变异(I145N),并且第151位氨基酸出现缺失情况;2016年潜在抗原位点第151位氨基酸也出现缺失情况,潜在抗原位点发生变异(Y155H)。
2.4.2 酶活性中心及周围相关位点的氨基酸序列分析 NA蛋白的酶催化活性位点包括催化部位的第118、151、152、224、276、292、371和406位等8个位点,除此还有酶活性中心周围的辅助位点11个,分别是第119、156、178、179、198、222、227、274、277、294和425位[4]。通过软件比对,发现2009年至2016年354条甲型H1N1流感病毒的NA基因所编码氨基酸在所有A型流感病毒的NA蛋白中都是保守的,未发现氨基酸位点突变。
2.4.3 糖基化位点的氨基酸序列分析 甲型H1N1流感病毒NA蛋白分子糖基化位点有8个,分别是第50、58、63、68、88、146、235和386位[3-4]。通过软件比对,发现2009至2016年354条甲型H1N1流感病毒的NA基因所编码的氨基酸在第50、58、63、68、88、146、235位未发生变异,保持稳定,但在第386位逐年变异,直至2016年该位点消失。
2.4.4 耐药性分析 NA某些氨基酸残基的突变会使病毒产生对这类抗流感病毒药物的耐药性,包括E119V、R292K、 R293K、N294S和H274Y位点[7-8],2009至2016年354条毒株序列尚未发现上述位点的突变, 表明对NA抑制剂仍敏感。
2.5 选择性压力分析 对2009至2016年各年份的NA序列分别采用SLAC、IFEL和REL模型进行选择性压力分析。各年份通过SLAC模型均未发现正向选择压力位点。IFEL模型发现2009年的序列有正向选择压力位点64;2010年有正向选择压力位点82和264;2013年有正向选择压力位点34、275和321;2016年有正向选择压力位点275,其余年份未发现。除2012年的序列外,通过REL模型均得到正向选择压力位点,结果见表2。
表2 中国2009至2016年甲型H1N1流感毒株正向选择压力位点分布

年份SLACIFELREL2009N/D6464、3912010N/D82、26462、82、3862011N/DN/D145、1792012N/DN/DN/D2013N/D34、275、32134、44、286、3092014N/DN/D144、2892015N/DN/D34、40、314、3462016N/D27534、40、93、96、257、275、314、396
注:N/D表示未发现。
2.6 进化动力学分析 Bayesian skyline reconstruction建立的模型表明,流感病毒遗传多样性在2009年维持稳定状态,并且在2009年和2010年达到高峰,随后分别在2011年及2012年有2次下降趋势,2012年6月呈现上升趋势,于2013年和2014年再次达到流行高峰,此后至2015年下降并维持于相对稳定状态,2016年开始又呈现上升趋势,预测2017年可能又将达到一个流行高峰。见图3。
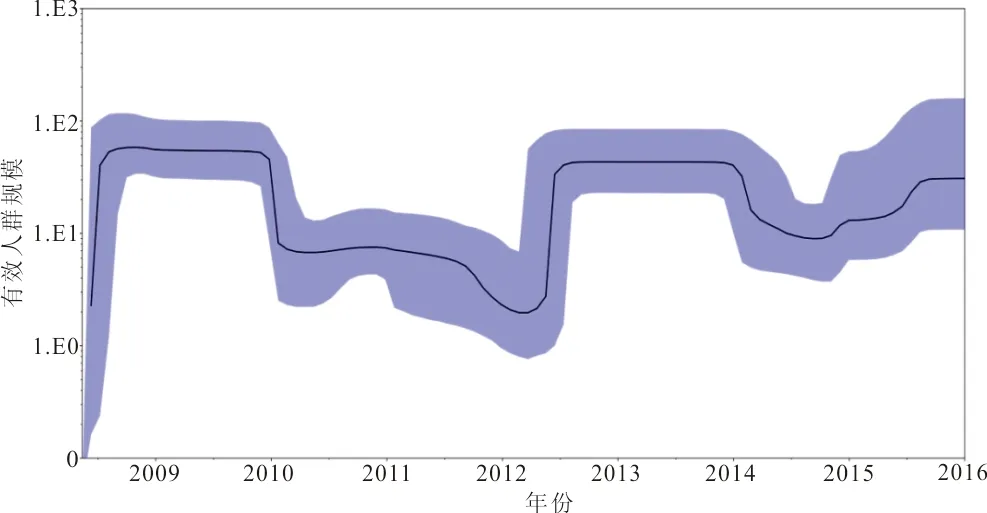
注:黑色实线为估计中位数,蓝色区域为95%置信区间。
图3 甲型H1N1流感NA基因进化动力学
3 讨论
根据国家流感中心的监测结果,2009至2016年中国流感流行主要以甲型H1N1、B型和季节性H3N2为主,其中甲型H1N1亚型作为我国流感流行的重要亚型,共发生4次流行高峰,主要在冬春季。2009至2016年甲型H1N1流感病毒NA基因与参考毒株A/California/07/2009(H1N1)的同源性逐年降低,类似研究也提示甲型H1N1流感病毒逐渐进化变异[9]。本研究发现NA有16个氨基酸位点改变,分别为V13I、I34V、N44S、V67I、N200S、V241I、N248D、V264I、N270K、I314M、I321V、I329N、N369K、T381I、N386K、K432E,其他变异位点呈散发型。核苷酸与氨基酸的遗传距离随着年份的增加而不断增大,遗传进化速度加快。流感病毒NA所编码的氨基酸关键位点突变增多。抗原位点的突变、糖基化位点的消失、酶活性位点的变异等都可能会影响流感病毒的抗原性和致病力[10],从而使病毒适应宿主感染环境,在人群中更广泛地传播。NA蛋白的酶活性中心位于NA的头部[11],可以直接或间接参与酶的催化作用。2009至2016年甲型H1N1流感病毒NA蛋白分子酶活性中心及周围相关位点相对保守,有利于稳定酶的活性及病毒扩散能力。NA催化活性位点可以直接或间接参与NA的催化功能,如有氨基酸变异将会影响NA的活性[12],NA抑制剂设计的原则就是基于NA高度保守的酶活性位点[13-14]。本研究并未发现酶活性位点或耐药位点的变异,表明相关位点仍可作为抗流感药物研发的基因靶点[11,15]。糖基化位点是维持NA蛋白分子结构和正常生物功能的重要影响因素,糖基化位点的变异对稳定病毒蛋白结构和生物特性有很大影响[16]。2009年新型甲型H1N1流感病毒具有8个糖基化位点,其中N386K随着年份的递增,直至彻底消失。研究表明,糖基化位点的非糖基化以及潜在糖基化位点数量的变化都可能会影响蛋白质折叠和运输等特性和功能,进而导致流感病毒结合受体的能力和病毒致病性降低[17]。病毒在进化过程中会获得新的糖基化位点,新的糖基化位点可能会掩盖抗原位点,成为病毒免疫逃逸的一种方式,需要加强监测。正向选择位点与病毒受体结合能力有关,因此这些正向位点的选择可能有助于H1N1流感的宿主从猪到人的适应。正向选择位点常发生于免疫水平较低的人群,会影响疫苗的近期效果评价[18]。
Ebranati等[19]认为病毒遗传多样性的波动与其所引起的暴发流行相对应。进化动力学趋势显示,甲型H1N1流感的流行趋势呈阶段性,每流行2年会间隔一年,与类似研究结果一致[20]。其中趋势图下降可能由于流感暴发后,国家采取了及时的公共卫生措施,从而控制了流感疫情,趋势图的上升可能由于病毒在自然界的不断变异,从而逃避宿主免疫引起流感的流行。
综上所述,随着流感病毒在人群中的进一步适应和持续存在,病毒分子特征将发生变异,预示该亚型在未来的时间里很有可能继续成为优势毒株。本研究追踪甲型H1N1流感病毒的进化趋势,特别关注基因变化对病毒的传播力和致病性的影响,从而及时发现具有流行病学意义的变异株和耐药株,这对疾病防控及预测具有重要的意义。
[1]韩一芳, 谢佳新, 殷建华, 等. 2009年新型甲型H1N1流感病毒基质蛋白及核蛋白基因进化分析[J]. 第二军医大学学报, 2009, 30(6): 622-627.
[2]方斌, 刘琳琳, 李翔, 等. 湖北省大流行期和流行后期新甲型H1N1流感病毒基因特性分析 [J]. 中华疾病控制杂志, 2017, 21(1): 13-18.
[3]赵红玲, 章文, 黄涛. 甲型H1N1流感病毒基因特征及耐药性研究[J]. 中国病原生物学杂志, 2014,9(9): 811-815.
[4]苏彤, 李淑华, 常文军, 等. 2009年新型甲型H1N1流感病毒神经氨酸酶基因进化分析[J]. 第二军医大学学报, 2009, 30(6): 618-621.
[5]Koel B F, Mogling R, Chutinimitkul S,etal. Identification of amino acid substitutions supporting antigenic change of influenza A(H1N1)pdm09 viruses [J]. J Virol, 2015, 89(7): 3763-3775.
[6]王杨,崔大伟,黄明珠,等.新型甲型H7N9禽流感病毒HA与NA的进化分析[J].临床检验杂志,2013,31(7):499-502.
[7]卢桂兰, 彭晓旻, 刘医萌, 等. 北京市2010年-2011年儿童甲型H3N2流感病毒神经氨酸酶基因进化和耐药性位点分析[J]. 中国卫生检验杂志, 2012,22(6): 1232-1236.
[8]祁贤, 汤奋扬, 李亮, 等. 新甲型H1N1(2009)流感病毒的早期分子特征[J]. 微生物学报, 2010,50(1): 81-90.
[9]颜文娟, 郭艳, 李小杉, 等. 2009-2015年中国甲型H1N1流感病毒血凝素基因进化分析[J]. 微生物学通报, 2017, 44(2): 420-427.
[10]Khandaker I, Suzuki A, Kamigaki T,etal. Molecular evolution of the hemagglutinin and neuraminidase genes of pandemic (H1N1) 2009 influenza viruses in Sendai, Japan, during 2009-2011 [J]. Virus Genes, 2013,47(3): 456-466.
[11]张建国, 张继宁, 黄勋娟. 神经氨酸酶抑制剂的研究进展[J]. 化学与生物工程, 2014, 31(4): 1-5,23.
[12]吴英萍,崔大伟,秦志梅,等. 2009~2012年杭州地区甲型H1N1流感病毒HA与NA基因的进化分析[J]. 临床检验杂志,2013,31(4):272-275.
[13]Richard M, Deleage C, Barthelemy M,etal. Impact of influenza A virus neuraminidase mutations on the stability, activity, and sensibility of the neuraminidase to neuraminidase inhibitors [J]. J Clin Virol, 2008, 41(1): 20-24.
[14]陈寅,茅海燕,周敏,等. 浙江省近年甲型H1N1流感病毒神经氨酸酶基因序列分析[J].疾病监测,2013,28(10):811-814.
[15]杨先知,崔大伟,谢国良,等.杭州市2013-2014年流感暴发流行期甲型流感病毒H3N2亚型基因特性分析[J].临床检验杂志,2015,33(8):586-590.
[16]谢剑锋, 沈晓娜, 王美爱, 等. 福建省甲型H1N1流感病毒基因特征研究[J]. 病毒学报, 2014, 30(1): 37-43.
[17]李向忠,方芳,陈则. 流感病毒神经氨酸酶不同区域的作用[J]. 生命科学研究, 2005, 9(2): 59-65.
[18]Smith DJ, Lapedes AS, De Jong JC,etal. Mapping the antigenic and genetic evolution of influenza virus[J]. Science, 2004, 305(5682): 371-376.
[19]Ebranati E, Pariani E, Piralla A,etal. Reconstruction of the evolutionary dynamics of A(H3N2) influenza viruses circulating in Italy from 2004 to 2012[J]. PLoS One, 2015, 10(9): e0137099.
[20]刘丹, 王建红, 陈洪永, 等. 2009年-2015年唐山地区流感病毒病原学特征[J]. 中国卫生检验杂志, 2017, 27(2): 185-187.
(本文编辑:刘群)
Evolution analysis and trend prediction of neuraminidase gene of influenza A/H1N1(09pdm) virus in China from 2009 to 2016
SONGYue1,YANWen-juan1,FANGKun1,LIXiao-shan1,LIWei2,WEIPing-min1
(1.SchoolofPublicHealth,South-EastUniversity,Nanjing210009,Jiangsu; 2.InfectiousDiseaseControlandSchoolHealth,NanjingCenterforDiseaseControlandPrevention,Nanjing210009,Jiangsu,China)
Objective To analyze the evolutionary characteristics and predict the variation trend of neuraminidase (NA) gene of influenza A/H1N1 (09pdm) virus in China from 2009 to 2016 in order to provide the basis of assessment for flu vaccines. Methods A total of 1 141 sequences of NA gene of influenza A/H1N1 (09pdm) virus were screened out from the Global Initiative on Sharing All Influenza Data and National Center for Biotechnology Information. The phylogenetic trees and the mutations of amino acids sequences were constructed and analyzed by biological softwares. The prediction for epidemic trend of influenza was analyzed by Bayesian skyline Plot. Results Compared with the sequence of reference strain, the homology of nucleic acid sequence of NA gene decreased year by year from 2009 to 2016. The phylogenetic analysis showed that NA gene clustered nearly on the identical phylogenetic tree in one year. The positive selection pressure site of NA strain was observed by different models in each year except 2012. The dynamics analysis showed that the popularity of influenza A/H1N1 virus may continue to increase to a peak in 2017. Conclusion The amino acid encoded by NA gene of influenza A/H1N1 virus is varying gradually, so the importance of surveillance for influenza virus should be reinforced for every year.
influenza A/H1N1(09pdm); neuraminidase; molecular characteristics; evolution analysis
10.13602/j.cnki.jcls.2017.07.19
江苏省重点研发计划(社会发展)项目(BE2015679);江苏省“六大人才高峰”第十一批高层次人才选拔培养资助项目(2014-WSN-40)。
宋玥,1992年生,女,硕士研究生,研究方向:流行病与卫生统计学。
卫平民,教授,博士,E-mail: mpw1963@126.com。
R373.1+3
A
2017-03-29)
猜你喜欢
科学大观园(2022年2期)2022-01-23
现代畜牧科技(2021年6期)2021-07-16
广东医科大学学报(2020年6期)2020-02-06
猪业科学(2018年6期)2018-01-23
中国猪业(2017年11期)2017-12-11
现代检验医学杂志(2016年3期)2016-11-15
现代检验医学杂志(2016年1期)2016-11-12
医学研究杂志(2015年12期)2015-06-10
特产研究(2014年4期)2014-04-10
郑州大学学报(理学版)(2014年3期)2014-03-01
