
镉导致高血压发生机制的研究进展
2017-08-16 10:23:11季星岐韩邦兴陈乃富施海峰
生物学杂志 2017年4期
季星岐, 韩邦兴, 陈乃富, 周 阳, 施海峰
(1. 江苏大学 生命科学研究院, 镇江 212013;2. 皖西学院 生物与制药工程学院 中药研究与开发工程技术研究中心, 六安 237012)
镉导致高血压发生机制的研究进展
季星岐1, 韩邦兴2, 陈乃富2, 周 阳1, 施海峰1
(1. 江苏大学 生命科学研究院, 镇江 212013;2. 皖西学院 生物与制药工程学院 中药研究与开发工程技术研究中心, 六安 237012)
镉具有很强的生物毒性,它可通过吸烟、饮食、呼吸等方式进入人体,并在人体内积累,造成机体损伤。镉在人体内积累后不但能导致骨骼畸形、肾脏损伤和癌症的发生,而且还能导致机体脂代谢紊乱,而脂代谢紊乱会导致心脑血管疾病的发生。近年来,有越来越多的研究结果发现镉能诱导哺乳动物产生心脏病、高血压等疾病,其分子机制包括镉产生大量自由基、镉抑制Na+/K+泵活性、镉影响VE-cadherin的功能等。综述了镉导致心脑血管疾病发生机制的研究进展。
镉;心脑血管疾病;VE-cadherin;自由基;Na+/K+泵
镉是人体生理活动和新陈代谢中非必需的金属元素,具有很强的生物毒性,在人体内积累后十分容易造成机体损伤。近年来发现镉能诱导心脑血管疾病的发生,其分子机制的研究也越来越受到关注。总结近几年的研究结果,镉诱导心脑血管疾病发生的机制有以下几个方面:1)镉产生氧化压力(Oxidative stress)使血浆中脂类过氧化,从而导致高血压的发生。镉是一种能对细胞造成很强氧化压力的重金属,镉进入血液后,将血浆中低密度脂蛋白(Low-density lipoprotein,LDL)过氧化,而被氧化修饰的LDL(Oxidized low-density lipoprotein,oxLDL)能在血管中大量积累,并且在血管壁产生斑块,诱发血管动脉粥样硬化。2)镉抑制Na+/K+泵(Na+/K+ATPase)活性,使肾小管细胞发生钠潴留而导致高血压。肾脏是镉在人体中积累最多的器官,镉在肾小管中积累后,会抑制肾小管细胞Na+/K+泵活性,导致细胞内的钠输出产生障碍,使得细胞内钠浓度升高。细胞内钠浓度升高后会引发细胞钠潴留,血管血容量增加,最终产生高血压。3)镉与钙竞争血管内皮细胞钙黏蛋白(Vascular endothelial cadherin,VE-cadherin)膜外钙离子结合位点,使血管通透性下降而导致高血压。VE-cadherin在调控血管内皮细胞通透性的过程中具有重要作用,镉能与钙竞争性结合VE-cadherin的膜外钙离子结合位点,导致血管内皮细胞的形变能力下降。同时镉还能降低VE-cadherin与血管内皮细胞内结构蛋白复合体之间的联系,使血管通透性下降、血管脆化,并最终引发高血压。
1 镉产生氧化压力导致高血压
1.1 镉导致氧化压力产生的机制
氧化压力为机体活性氧成分(Reactive oxygen species,ROS)与抗氧化系统之间平衡失调引起的一系列适应性的反应。当细胞正常的氧化还原状态被干扰后,细胞会制造出过氧化物与自由基,这些物质会对细胞产生毒性,损害细胞的蛋白质、脂类和DNA。镉导致氧化压力产生的机制主要有以下几个方面:1)镉通过间接方式引发芬顿反应(Fenton Reaction)产生氧化压力。细胞质中和细胞膜上有很多与铁、铜结合的蛋白(如铁蛋白,铜蓝蛋白),镉能取代铁、铜与这些蛋白结合,导致细胞中游离的铁离子和铜离子浓度增加。这些游离的金属离子能引发Fenton Reaction产生超氧自由基、羟基自由基和氮氧自由基[1]。2)镉结合SOD和GSH产生氧化压力。超氧化物歧化酶(Superoxide dismutase,SOD)和谷胱甘肽(Glutathione,GSH)氧化还原系统是人体中主要的抗氧化剂,它们都富含巯基,而镉对SOD和GSH的巯基有很高的亲和力,当镉和SOD和GSH的巯基结合后会导致细胞内氧化还原稳态失衡,从而导致氧化压力的产生[2-4]。3)镉改变线粒体结构和功能产生氧化压力[5-6]。镉能作用于线粒体呼吸链中的复合体Ⅲ,使线粒体渗透转换孔(Mitochondrial permeability transition pore,MPTP)打开,从而导致线粒体产生的ROS释放到细胞质中。同时,镉还能导致线粒体中不稳定半泛醌(Semiubiquinones)的积累,使其易于向分子氧转移一个电子,从而导致过氧化物的产生[7]。
1.2 镉导致血浆脂类过氧化和斑块形成
镉诱导产生的自由基使血浆中脂类产生过氧化反应,从而导致心脑血管损伤[8]。LDL被氧化后形成的oxLDL可能是导致动脉粥样硬化的原因[9]。
在血管中,LDL中的长链多不饱和脂肪酸会被血管内皮细胞、平滑肌细胞和巨噬细胞产生的ROS氧化修饰,产生共轭双烯和脂质过氧自由基(LOO-)[10]。LOO-会攻击相邻的脂肪酸分子,并使其分解。同时,LDL中脂类被氧化后产生大量丙二醛(Malondialdehyde,MDA)和溶血磷脂(Lysophosphatide),并在LDL中积累。这些物质修饰载脂蛋白B100(Aplolipoprotein B100,ApoB100)的氨基酸链形成oxLDL[11]。oxLDL由于其氨基酸被氧化修饰后无法被LDL受体识别,最终会被巨噬细胞吞噬。通过这种方式,巨噬细胞内吞噬了大量oxLDL,使细胞内充满脂类并且聚集在血管内皮中,这种富含脂类物质的巨噬细胞在血管内皮中积累会导致血管中脂类无法被代谢,从而诱发血管动脉粥样硬化。同时,oxLDL的产生还会产生多种不良效应,如刺激单核细胞(Monocyte)释放大量TNF-α、IL-1β,而这些细胞因子会导致平滑肌细胞分化,产生大量胶原蛋白(Collagen)和弹性蛋白(Elastin),促进血管中斑块形成和血管纤维化[12]。另外,oxLDL还能抑制环前列腺素(Prostacyclin)的合成[12],而环前列腺素在阻遏血小板聚集上有重要作用,故其合成减少会导致血小板在血管中聚集。血小板聚集后能释放生长因子,导致平滑肌细胞的分化和迁移,同时还能诱导血栓的形成。
2 镉抑制Na+/K+泵活性导致高血压
2.1 Na+/K+泵结构和功能
Na+/K+泵是一种细胞膜蛋白,主要由α,β和γ 3个亚基组成[13]。α亚基为分子质量约120 ku的10次跨膜蛋白,它具有和Na+、K+结合的活性,同时又具有水解ATP产生能量的活性[14],α亚基还含有一个乌本苷结合位点(Ouabain-binding site),该位点是强心类固醇(Cardiotonic steroids)的受体[15];β亚基分子质量约为50 ku,其主要功能是维持Na+/K+泵在细胞膜上的稳定性;γ亚基有2个亚型,γ(a)和γ(b)。γ(a)主要在肾脏近曲小管上皮细胞中表达,并且调控Na+/K+泵活性[16]。
Na+/K+泵首先在膜内侧与细胞内的Na+结合,ATP酶活性被激活后,由ATP水解释放的能量使Na+/K+泵构象发生变化,将Na+输出细胞;与此同时,Na+/K+泵与细胞膜外侧的K+结合,发生去磷酸化后构象再次改变,将K+输入细胞内。研究表明,每消耗1个ATP分子,可使细胞内减少3个Na+并增加2个K+。
2.2 镉影响抑制细胞Na+/K+泵活性导致高血压
Na+/K+泵为Na+的重吸收提供能量,抑制Na+/K+泵会导致细胞钠潴留。在血管平滑肌细胞中,当Na+/K+泵活性被抑制后,细胞中Na+浓度升高,Na+/Ca2+交换所需的Na+浓度梯度随之降低,使得细胞内的Ca2+浓度上升,从而导致血管平滑肌细胞伸缩性增加,并且导致高血压。
在肾小管中积累的镉能通过蛋白酶体途径(Proteasomal degradation pathway)和溶酶体途径(Lysosomal degradation pathway)降解Na+/K+泵,从而抑制肾小管细胞排出Na+。细胞内Na+浓度升高后会引起肾小管钠潴留,血容量增加,从而导致高血压[17]。同时,细胞内的高钠又会导致Na+和Ca2+之间的交换减少,细胞内Ca2+浓度升高,血管平滑肌收缩,肌张力和外周阻力增高,最终引起高血压。
镉还能通过抑制多巴胺(Dopamine)的合成来影响Na+/K+泵的功能[18]。Dopamine通过和细胞表面多巴胺受体D1、D3结合来调控Na+/K+泵的活性,当Na+/K+泵活性过高时,Dopamine促进细胞内吞来抑制Na+/K+泵活性,从而调节细胞内Na+浓度[19]。镉能增加细胞Dopamine的合成,当多巴胺的含量增加时,能抑制肾小管细胞Na+/K+泵的功能,从而导致肾小管细胞排钠量降低,细胞内发生钠潴留,最终引起高血压。
血管紧张素Ⅱ(Angiotensin II)在肾小管中能通过血管紧张素Ⅱ受体信号通路(AT1R signaling pathway)调控Na+的重吸收。低浓度的血管紧张素Ⅱ增加Na+/K+泵活性;高浓度抑制其活性[20]。镉能导致血管紧张素Ⅱ的合成增加,导致Na+/K+泵活性被抑制,引起细胞内Na+潴留。同时,镉能增加血管紧张素转换酶(Angiotensin-converting enzyme)的活性,导致血管紧张素Ⅱ的合成增加,促进前列腺素的合成(Prostanoid)[21],前列腺素具有调节血管紧张性的作用,过量合成的前列腺素会导致血管收缩性增加,导致高血压[22]。
3 镉影响VE-cadherin导致高血压
3.1 VE-cadherin的结构和功能
VE-cadherin是一种在内皮细胞表面特异性表达的黏附蛋白,它在维持内皮细胞之间的黏附上起着重要的作用。在血管内皮细胞中,VE-cadherin能调控血管通透性和白细胞外溢,并且能调节细胞分化、凋亡和内皮细胞表面生长因子受体的表达[23]。
VE-cadherin包含5个胞外cadherin结构域(EC domain),EC domain和Ca2+的结合是VE-cadherin发挥细胞黏附作用的重要条件[23]。VE-cadherin的C端胞内区主要和细胞质中的catenins结合,VE-cadherin和β-catenin结合后,β-catenin会和α-catenin结合,α-catenin又和肌动蛋白actin相结合,它们形成的VE-cadherin-β-catenin-α-catenin-actin复合体在维持细胞结构和细胞之间的黏附上具有重要的作用[23]。
VE-cadherin能调控血管内皮细胞通透性[24]。血管内皮是沿着血管内侧生长、调节液体和溶质在血液和周围组织之间流动的半渗透屏障,它将血管与周围的组织隔开并控制着两者之间的物质运输,这种控制物质运输的能力即为血管内皮的通透性。将VE-cadherin和β-catenin在细胞内的结合区进行突变后,细胞间的连接能力显著下降,并且还会引起血管形变。向成年小鼠体内注入抗VE-cadherin的抗体后会导致小鼠血管内皮细胞通透性下降、血管脆化并出血[25]。这些研究表明,VE-cadherin能调控内皮细胞的通透性和细胞间的稳定性。
3.2 镉与钙竞争VE-cadherin钙离子结合位点
VE-cadherin和高血压的发生密切相关[26]。镉作为一种非生理必需的重金属,会和VE-cadherin特异性结合而影响其正常的生理功能,从而导致血管通透性减弱、炎症反应和高血压等疾病的发生。
VE-cadherin的膜外区上有Ca2+结合位点,当它和Ca2+结合后,其构象会变得具有刚性,两个相邻细胞的VE-cadherin就能彼此接触并连接到一起。细胞外Ca2+被EGTA螯合后,VE-cadherin的构象会发生变化,并且导致细胞无法黏附[25]。研究表明,镉能抑制VE-cadherin在维持细胞形态中的作用。5~100 μmol/L CdCl2处理Caco-2细胞4 h后细胞的通透性增加,并且cadherin的胞内区与catenin结合位点发生了改变[27]。在镉诱导肺损伤的小鼠模型中,镉暴露小鼠血管内皮细胞中的VE-cadherin显著下降[28]。另外,体外CdCl2处理VE-cadherin膜外EC domain,圆二色谱(Circular dichroism)检测发现Cd2+能导致该肽段的二级结构发生显著变化。这些研究表明,镉能和cadherin的EC domain结合,导致Ca2+无法和cadherin结合,造成cadherin黏附细胞功能丧失。因此,镉诱导的VE-cadherin结构的改变是诱发动脉粥样硬化和心脑血管疾病的重要因素。
4 镉导致高血压的其他机制
镉还能导致血清中脂类水平发生显著变化。给大鼠喂养含镉浓度为1 mg/kg的食物,持续喂养4周,采血后检测其中脂类物质,发现总胆固醇(Total cholesterol,TC)、低密度脂蛋白胆固醇(Low-density lipoprotein cholesterol,LDL-C)和甘油三酯(Triglyceride,TG)含量显著上升[29]。另一项研究表明,给大鼠喂养含镉浓度为3 mg/kg的食物,持续喂养21 d后检测血浆脂类含量,发现TC、极低密度脂蛋白胆固醇(Very low-density lipoprotein cholesterol,VLDL-C)、LDL-C、TG、脂肪酸(Fatty acid,FA)和磷脂(Phospholipids)都显著上升。这些脂类物质在血浆中含量上升会导致血管中斑块的形成,使动脉越来越窄,引起心绞痛,一旦溃破还会形成血栓,堵塞动脉,导致心肌梗死、中风等严重问题。这些研究表明镉能通过直接影响血清中脂类物质的代谢过程来引发心脑血管疾病,然而其分子机制还不是十分清楚,因此镉如何导致血清中脂类物质代谢异常还有待解决(图1)。
5 结论与展望
镉具有很强的生物毒性,并且其半衰期很长,一旦被摄入人体就很难被代谢,从而对人体各组织和器官造成损伤。镉离子进入细胞后能间接产生自由基,会影响一系列细胞信号的传导,甚至导致细胞凋亡。除了产生ROS外,镉还能参与细胞的钙离子代谢过程,从而影响细胞对钙的吸收和排出。另外,镉还能抑制自然杀伤细胞(Natural killer cell,NK)的活性,降低机体的免疫力。总而言之,镉能多方位对生物机体产生毒性,造成严重的健康问题。如20世纪初发生在日本富山县的骨痛病,就是由于在该地区的人饮用含有大量镉的水,使得镉在人体中积累,从而导致病人骨质疏松、骨骼萎缩和关节疼痛。
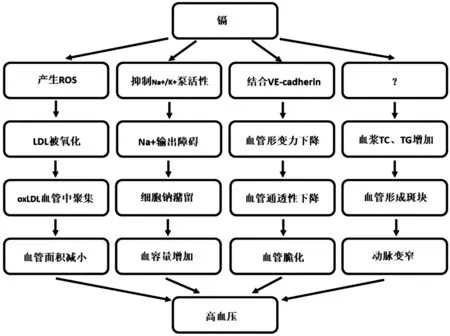
图1 镉诱导高血压发生的分子机制
在一些发展中国家,镉污染现象十分严重,并且随着工业化进程的加快,其现状越来越恶化,因此,镉污染的防治就变得尤其重要。目前,行之有效的镉污染防治措施包括污染土壤的物理隔离;控制锌矿过度开采;严格按照排放标准检测废水、废气中镉含量等。然而,这些措施虽然能在一定程度上控制镉污染现象的发生,但是对于有关行业的发展具有很大的负面作用。因此,能在兼顾发展的同时又能有效控制镉污染就变得尤为重要了。在生物学飞速发展的今天,运用生物学方法对镉污染进行有效地检测和预防给人们带来了新的思路。目前世界上已有很多实验室把目光聚焦于镉导致机体各种损伤的分子机制上,试图从分子机制层面解释镉的生物毒性,并且已经在某些方面清晰地阐明了镉诱导机体损伤的分子机制。然而,对于近年来发现的镉诱导产生心脏病、高血压等心脑血管疾病现象的分子机制的报道还不够完善,鉴于其对人体健康的重要性而言,尽快阐明镉引起心脑血管疾病的分子机制无疑是造福人类的一项巨大贡献。
[1]RECZEK C R,CHANDEL N S. ROS-dependent signal transduction [J]. Current Opinion in Cell Biology, 2015, 33: 8-13.
[2]JOMOVA K,VALKO M. Advances in metal-induced oxidative stress and human disease [J]. Toxicology, 2011, 283(2): 65-87.
[3]KASPERCZYK A, MACHNIK G, DOBRAKOWSKI M, et al. Gene expression and activity of antioxidant enzymes in the blood cells of workers who were occupationally exposed to lead [J]. Toxicology, 2012, 301(1-3): 79-84.
[4]NAIR A R, DEGHESELLE O, SMEETS K, et al. Cadmium-induced pathologies: where is the oxidative balance lost (or not)? [J]. International Journal of Molecular Sciences, 2013, 14(3): 6116-6143.
[5]FEDERICO A, CARDAIOLI E, DA POZZO P, et al. Mitochondria, oxidative stress and neurodegeneration [J]. Journal of the Neurological Sciences, 2012, 322(1-2): 254-262.
[6]PATHAK N, KHANDELWAL S. Oxidative stress and apoptotic changes in murine splenocytes exposed to cadmium [J]. Toxicology, 2006, 220(1): 26-36.
[7]ARROYO V, FLORES K, ORTIZ L, et al. Liver and cadmium toxicity [J]. J Drug Metab Toxicol S, 2012, 5(1): 32-37.
[8]OLISEKODIAKA M J, IGBENEGHU C A, ONUEGBU A J, et al. Lipid, lipoproteins, total antioxidant status and organ changes in rats administered high doses of cadmium chloride [J]. Medical Principles and Practice, 2011, 21(2): 156-159.
[9]PIRILLO A, NORATA G D,CATAPANO A L. LOX-1, OxLDL, and atherosclerosis [J]. Mediators of Inflammation, 2013: 152786.
[10]FUTEMA M, SHAH S, COOPER J A, et al. Refinement of variant selection for the LDL cholesterol genetic risk score in the diagnosis of the polygenic form of clinical familial hypercholesterolemia and replication in samples from 6 countries [J]. Clinical Chemistry, 2015, 61(1): 231-238.
[11]SINGH R, DEVI S,GOLLEN R. Role of free radical in atherosclerosis, diabetes and dyslipidaemia: larger‐than‐life [J]. Diabetes/Metabolism Research and Reviews, 2015, 31(2): 113-126.
[12]LIBBY P, RIDKER P M,HANSSON G K. Progress and challenges in translating the biology of atherosclerosis [J]. Nature, 2011, 473(7347): 317-325.
[13]MORTH J P, PEDERSEN B P, BUCH-PEDERSEN M J, et al. A structural overview of the plasma membrane Na+, K+-ATPase and H+-ATPase ion pumps [J]. Nature Reviews Molecular Cell Biology, 2011, 12(1): 60-70.
[14]IP Y K, LOONG A M, KUAH J S, et al. Roles of three branchial Na+-K+-ATPase α-subunit isoforms in freshwater adaptation, seawater acclimation, and active ammonia excretion inAnabastestudineus[J]. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology, 2012, 303(1): R112-R125.
[15]MIJATOVIC T,KISS R. Cardiotonic steroids-mediated Na+/K+-ATPase targeting could circumvent various chemoresistance pathways [J]. Planta Medica, 2013, 79(3-4): 189-198.
[16]YATIME L, LAURSEN M, MORTH J P, et al. Structural insights into the high affinity binding of cardiotonic steroids to the Na+, K+-ATPase [J]. Journal of Structural Biology, 2011, 174(2): 296-306.
[17]WILLIAMS B, MACDONALD T M, MORANT S, et al. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomised, double-blind, crossover trial [J]. The Lancet, 2015, 386(10008): 2059-2068.
[18]ROUSHANI M, SHAMSIPUR M, RAJABI H R. Highly selective detection of dopamine in the presence of ascorbic acid and uric acid using thioglycolic acid capped CdTe quantum dots modified electrode [J]. Journal of Electroanalytical Chemistry, 2014, 712(1):19-24.
[19]ZHANG L N, LI J X, HAO L, et al. Crosstalk between dopamine receptors and the Na+/K+-ATPase (Review) [J]. Molecular Medicine Reports, 2013, 8(5): 1291-1299.
[20]FIGTREE G A, KARIMI G K, LIU C C, et al. Oxidative regulation of the Na+-K+pump in the cardiovascular system [J]. Free Radical Biology and Medicine, 2012, 53(12): 2263-2268.
[21]WESELER A R, BAST A. Oxidative stress and vascular function: implications for pharmacologic treatments [J]. Current Hypertension Reports, 2010, 12(3): 154-161.
[22]HU Z W, KERB R, SHI X Y, et al. Angiotensin II increases expression of cyclooxygenase-2: implications for the function of vascular smooth muscle cells [J]. Journal of Pharmacology and Experimental Therapeutics, 2002, 303(2): 563-573.
[23]VESTWEBER D. VE-cadherin the major endothelial adhesion molecule controlling cellular junctions and blood vessel formation [J]. Arteriosclerosis, Thrombosis, and Vascular Biology, 2008, 28(2): 223-232.
[24]TOZER G, KANTHOU C, LEWIS G, et al. Tumour vascular disrupting agents: combating treatment resistance [J]. The British Journal of Radiology, 2008, 81: S12-S20.
[25]SCHULTE D, KÜPPERS V, DARTSCH N, et al. Stabilizing the VE-cadherin-catenin complex blocks leukocyte extravasation and vascular permeability [J]. The EMBO Journal, 2011, 30(20): 4157-4170.
[26]NIKITOPOULOU I, ORFANOS S E, KOTANIDOU A, et al. Vascular endothelial cadherin downregulation as a feature of endothelial transdifferentiation in monocrotaline-induced pulmonary hypertension [J]. American Journal of Physiology-Lung Cellular and Molecular Physiology, 2016, 311(2):L352-L363.
[27]FORTI E, BULGHERONI A, CETIN Y, et al. Characterisation of cadmium chloride induced molecular and functional alterations in airway epithelial cells [J]. Cellular Physiology and Biochemistry, 2009, 25(1): 159-168.
[28]HELMESTAM M, STAVREUS-EVERS A,OLOVSSON M. Cadmium chloride alters mRNA levels of angiogenesis related genes in primary human endometrial endothelial cells grown in vitro [J]. Reproductive Toxicology, 2010, 30(3): 370-376.
[29]REN X M, WANG G G, XU D Q, et al. The protection of selenium on cadmium-induced inhibition of spermatogenesis via activating testosterone synthesis in mice [J]. Food and Chemical Toxicology, 2012, 50(10): 3521-3529.
Research progress in mechanisms of cadmium induced hypertension
JI Xing-qi1, HAN Bang-xing2, CHEN Nai-fu2, ZHOU Yang1, SHI Hai-feng1
(1. Institute of Life Sciences, Jiangsu University, Zhenjiang 212013; 2. Engineering Technology Research Center of Research and Development of Traditional Chinese Medicine, College of Biological and Pharmaceutical Engineering, West Anhui University, Lu′an 237012, China)
Cadmium has strong biological toxicity. It could be absorbed by smoking, dietary and inhalation. Cadmium could lead to series damage to mammalian, including bone misshapen, kidney injury and cancer; it can also lead to abnormal lipid metabolism. In recent years, many researchers found that cadmium could lead to cardiovescular diseases in mammalians, such as heart attack, hypertension and so on,however, the mechanism is still unknown. The mechanism of this phenomenon could be the result of ROS(Reactive oxygen species) production, Na+/K+ATPase activity inhibition and impaired VE-cadherin function. Here, we discussed the research progress in mechanisms of cadmium induced hypertension.
cadmium; cardiovescular diseases; VE-cadherin; ROS; Na+/K+ATPase
2016-08-01;
2016-08-15
国家自然科学基金项目(No.31271272、31301919);江苏省自然科学基金(BK20130506);安徽省自然科学基金(1508085MH203);安徽省高等学校省级自然科学基金项目(KJ2011A270、KJ2013A265);江苏省六大人才高峰项目“人细胞铁代谢紊乱与人的相关疾病”
季星岐,硕士研究生,生物化学与分子生物学,E-mali:olivia_jxq@sina.com
施海峰,教授,博士生导师,研究方向为铁代谢、重金属毒性, E-mail:shihf@ujs.edu.cn
R544.1
A
2095-1736(2017)04-0094-04
doi∶10.3969/j.issn.2095-1736.2017.04.094
猜你喜欢
现代临床医学(2023年1期)2023-03-24 08:30:20
中老年保健(2021年7期)2021-08-22 07:42:04
今日农业(2020年23期)2020-12-15 03:48:26
广东医科大学学报(2020年6期)2020-02-06 06:00:38
广东茶业(2019年1期)2019-04-28 08:32:30
基层中医药(2018年4期)2018-08-29 01:25:56
中国医药指南(2017年3期)2017-11-13 02:55:49
癌变·畸变·突变(2016年5期)2016-08-22 05:55:18
东方考古(2016年0期)2016-07-31 17:45:44
畜牧兽医学报(2015年3期)2015-07-05 08:22:42
