
钾通道Kv4.2、Kv1.4在硫化氢后处理对大鼠短暂全脑缺血神经保护中的作用研究
2017-08-16 09:39:45拜承萍赵晨亮
中风与神经疾病杂志 2017年7期
拜承萍, 赵晨亮
钾通道Kv4.2、Kv1.4在硫化氢后处理对大鼠短暂全脑缺血神经保护中的作用研究
拜承萍1, 赵晨亮2
目的 研究硫氢化钠(sodium hydrosulfide,NaHS)后处理对短暂全脑缺血大鼠海马中钾通道Kv4.2和Kv1.4 mRNA表达变化的影响及其脑保护作用,从而探讨NaHS对大鼠短暂全脑缺血神经保护作用的机制。方法 用4VO方法建立大鼠短暂性全脑缺血(transient global cerebral ischemia,tGCI)模型,大鼠被随机分配到3组,分别为:假手术组(sham)、tGCI组、NaHS后处理组。NaHS后处理组为 tGCI之后1 d,给予大鼠腹腔注射NaHS 24 μmmol/kg或者180 μmmol/kg。通过尼氏染色与NeuN免疫染色确定海马神经元的死亡,通过RT-PCR方法检测海马组织Kv4.2和Kv1.4 mRNA 水平的表达变化。结果 (1)与tGCI组比较,在tGCI之后1 d给予24 μmol/kg NaHS后处理使海马CA1区存活细胞数目显著增加,而高剂量的 NaHS(180 μmol/kg)后处理对tGCI大鼠海马CA1区则无明显的保护作用。(2)在Re 26 h和Re 48 h,海马组织中Kv4.2、Kv1.4的mRNA 表达水平均明显低于假手术组(P<0.05)。在Re 26 h+NaHS组,kv4.2(1.24±0.08)和kv1.4(1.11±0.07)的mRNA表达水平均分别高于Re 26 h组的kv4.2(0.75±0.04)和kv1.4(0.79±0.06),差异均有显著性(P<0.05)。结论 外源性NaHS可能通过上调大鼠tGCI后海马区Kv4.2和Kv1.4 mRNA的表达,从而导致膜电位超极化,降低神经元兴奋性和氧耗,继而保护神经元免受脑缺血损伤。
短暂性全脑缺血; 钾通道; 硫氢化钠; 神经保护
缺血性卒中是导致死亡和残疾的主要原因。后处理可在脑缺血发生后应用并减轻再灌注损伤,开展各种后处理方式的基础及临床研究,将有利于开发后处理“模拟”药物。此外,由于缺血后神经元损害的机制尚未完全清楚[1],对其有效治疗极为有限。兴奋毒性是缺血后神经元损伤的主要原因,研究认为A型钾电流IA对调节神经元的兴奋性具有重要作用,可能影响缺血后兴奋毒性所致的细胞坏死的发病机制[2]。Kv4.2、Kv1.4属于A型钾通道,它们是IA的主要促成者,2001年Huang等[3]的研究表明,钾通道基因表达水平的变化与脑缺血时钾电流发生的变化相一致。本研究采用NaHS作为一种化学药物后处理方式来诱导脑缺血后的神经保护,并研究在短暂全脑缺血模型中NaHS对钾离子通道Kv4.2、Kv1.4 mRNA表达变化的影响,初步探讨NaHS对大鼠短暂全脑缺血神经保护作用的机制。
1 材料与方法
1.1 材料 (1)仪器与设备:大鼠立体定位固定仪(C5G01-008,68001,深圳市瑞沃德生命科技有限公司);切片机(Leica,德国);大型冷冻离心机、PCR仪(Eppendorf,德国),ABI7500 荧光仪、81Biorad 凝胶成像系统均购自美国。(2)试剂、耗材:NaHS(Sigma公司,美国);尼氏染色液(索莱宝公司,北京);NEUN抗体(Merck Millipore,德国)。
1.2 实验方法
1.2.1 实验动物分组及给药 选用成年健康雄性Wistar大鼠,从北京维通利华购买[合格证号:SCXK(京)2012-0001],体重200~250 g。分组与给药:大鼠被随机分配到3组,分别为:假手术组(sham)、tGCI组、NaHS后处理组,NaHS后处理组为 tGCI之后1 d,给予大鼠腹腔注射NaHS 24 μmmol/kg或者180 μmmol/kg。本研究纳入实验的动物44只,其中24只大鼠用于形态学检测,于再灌注7 d取脑观察形态学变化;20只大鼠用于RT-PCR检测,分别在再灌注之后的26 h、48 h断头取脑。
1.2.2 全脑缺血模型的制备 10%水合氯醛(350 mg/kg Wt)腹腔注射麻醉。颈部正中横切口,分离两侧颈总动脉约1 cm,将缺血装置套上颈总动脉,即将一条硅胶管系于颈总动脉周围并将其穿过聚乙烯纽扣的两个小孔,并穿过一个2 cm长的塑胶圆筒,打结,避免血管受压和血流受阻,简易缝合切口。将大鼠固定于定位仪上,枕部纵切口,暴露第一颈椎板及横突板上的翼小孔,用双极电凝针电凝双侧翼小孔内的椎动脉,缝合切口后待动物完全苏醒,将其移至笼内饲养,夜间禁食,此时动物行为正常,没有明显的脑损伤。24 h后在大鼠完全清醒的状态下,用无齿小动脉夹夹闭双侧颈总动脉15 min,大鼠在缺血开始后30~60 s 内昏迷,翻正反射消失,能自主呼吸,双侧瞳孔放大,痛觉反射消失,这些症状持续于整个缺血过程中,凡未出现以上症状或这些症状未能持续整个缺血过程中的大鼠弃用。缺血15 min后解除夹闭的缺血装置,恢复脑血流,动物即逐渐恢复知觉开始活动,未见明显障碍,凡缺血后有癫痫、偏瘫、惊厥等症状的大鼠均弃用。
1.2.3 脑取材用于形态学观察的标本 麻醉动物后用4 ℃生理盐水100 ml 经心脏快速灌注冲净血液,随后用4 ℃ 的4%的多聚甲醛以先快后慢的速度灌注固定。取脑放入4 ℃的4%的多聚甲醛中进行后固定,并先后放入15%、30%的梯度蔗糖多聚甲醛溶液中脱水,OCT包埋,-20 ℃下进行冰冻切片,选取典型海马区脑片,一部分用于做尼氏染色,光镜下观察组织学特点,另一部分做NeuN免疫染色。
1.2.4 尼氏染色 1%焦油紫3 min→ 70%乙醇脱水1 min→ 80%乙醇脱水1 min →90%乙醇1 min→95%乙醇(加数滴冰醋酸)中数秒→100%乙醇Ⅰ脱水3 min→100%乙醇Ⅱ脱水3 min→二甲苯Ⅰ透明5 min→二甲苯Ⅱ透明5 min→ 中性树胶封片镜检。尼氏染色的结果进行阳性细胞计数:在高倍镜下(×200)选取3个不重复的视野进行阳性细胞计数后取平均值,每只动物计数2个脑片。
1.2.5 NeuN免疫染色 (1)PBS洗2~3次各5 min;(2)3%H2O2室温静置30 min;(3)PBS洗2~3次各5 min;(4)滴加正常山羊血清封闭液,室温60 min;(5)滴加Ⅰ抗NeuN(浓度1∶5000),4 ℃过夜;(6)PBS洗5次各5 min;(7)滴加Ⅱ抗室温静置2 h;(8)PBS洗3次各5 h;D液30 min,PBS洗5 min 3次;(9)DAB显色5~10 min;(10)脱水、透明、封片、镜检。
1.2.6 PCR实验过程 将麻醉的大鼠迅速断头取脑,在冰盒上分离出大脑海马,置于液氮保存。用Trizol 按产品所提供操作步骤提取组织总RNA。提取的RNA A260/A280在1.8~2.0之间,且琼脂糖凝胶电泳,28S和18S条带比值约为2∶1,说明所提取的RNA 无降解和污染。
1.2.6.1 反转录反应 在反应液中依次加入4 μl 5×PrimeScript RT Master Mix,稀释后的RNA,RNase Free dH2O 至20 μl 反应体系。然后置于PCR 仪37 ℃,15 min,85 ℃ 5 s,4 ℃之后置于-20 ℃保存。
1.2.6.2 Q-PCR反应 在96 孔板内按顺序依次加入SYBR Premix Ex TaqⅡ 10 μl,PCR Forward Primer 0.8 μl,PCR Reverse Primer 0.8 μl,DNA模板2 μl,dH2O 6.4 μl,Total 20 μl。于ABI7500 荧光仪设置stage1:95 ℃预变性30 s;Stage2:PCR反应,95 ℃ 5 s,60 ℃ 30 s,重复40 个循环后;stage3:95 ℃ 15 s,60 ℃ 60 s,95 ℃ 15 s。每个反应设3 个复孔。引物设计合成参照文献[4]中Kv1.4、Kv4.2的基因序列,由美国QIAGEN公司合成引物。数据分析:实验数据使用ABI 7500 自动分析软件得出Ct值,将Ct值作为样本表达量标准,采用β-actin作为内参照,采用2-△△Ct方法计算目的基因的相对表达水平,即目的基因表达差异=2-△△Ct,△△Ct = (CT-CTβ-actin)处理组-(CT-CTβ-actin) 对照组。
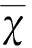
2 结 果
2.1 NaHS后处理对于tGCI诱导的海马CA1区迟发性神经元损害的效应 用尼氏染色和NeuN免疫染色方法在15 min的tGCI(有或没有NaHS后处理)之后7 d,评估海马CA1区神经元的丢失,如图1所示,假手术组大鼠显示没有组织病理学异常。在tGCI鼠的CA1区,我们观察到大片神经元损伤,表现为神经元的肿胀伴随尼氏体的丢失。相比较tGCI组,24 μmol/kg NaHS后处理显著增加了CA1区存活细胞的数目(可见淡染色的核和完整的尼氏体),通过NeuN染色我们获得了相似的结果。图2定量分析证实,和tGCI组相比,24 μmol/kg NaHS后处理组大鼠,存活的CA1锥体神经元平均数目显著升高(P<0.05),但同时我们发现,高剂量的 NaHS(180 μmol/kg)后处理未显示保护效应。
右边图片是对左侧CA1平台区域的放大。标尺:a-d:500 μm;e-h:200 μm;i-l:100 μm;m-p:50 μm。sham组:(a,e,i,m);tGCI组:15 min tGCI之后7 d观察组织形态学 (b,f,j,n);24 μmol/kg 和180 μmol/kg NaHS后处理组:15 min tGCI之后1 d腹腔注射NaHS,再灌注之后7 d观察组织形态学(c,g,k,o,d,h,l,p)
图1 tGCI之后7 d,大鼠海马的尼氏染色、NeuN染色的显微照片
2.2 Kv4.2 和Kv1.4 mRNA 在大鼠海马的表达水平 (1)Kv4.2 mRNA在大鼠海马的相对表达量 表1及图3示,与sham组相比,各tGCI组再灌注不同时间点的Kv4.2的mRNA表达水平均明显下调(P=0.001),与sham组相比,Re 26 h+NaHS组的表达明显上调(P=0.001),而Re 48 h+NaHS组的表达下调(P=0.004)。tGCI组Re 26 h与Re 26 h+NaHS组相比,NaHS组Kv4.2的mRNA表达水平明显上调(P=0.000),tGCI组Re 48 h与Re 48 h+NaHS组之间表达无明显差异(P=0.071);(2)Kv1.4 mRNA在大鼠海马的相对表达量 表2及图3示,与sham组相比,各tGCI组再灌注不同时间点的Kv1.4的mRNA表达水平均明显下调(P=0.001),与sham组相比,Re 26 h+NaHS组表达无明显差异(P=0.363),Re48h+NaHS组的表达下调(P=0.001)。tGCI组Re 26 h与Re 26 h+NaHS组相比,NaHS组Kv1.4的mRNA表达水平明显上调(P=0.003),tGCI组Re 48 h与Re 48 h+NaHS组之间表达无明显差异(P=0.263)。

图2 NaHS后处理对于海马CA1区神经元存活的效应
表1 Kv4.2在大鼠海马的mRNA相对表达量
与sham组比较,a:P<0.05;与tGCI组比较,b:P<0.05
表2 Kv1.4 在大鼠海马的 mRNA 相对表达量
与sham组比较,a:P<0.05;与tGCI组比较,b:P<0.05

图3 Kv4.2 和Kv1.4在大鼠海马的 mRNA 相对表达量
3 讨 论
长久以来,硫化氢(hydrogen sulfide,H2S)被认为是一种有毒物质,直到最近,研究发现低浓度的H2S 在正常生理过程中扮演着重要角色[5]。H2S 在星型胶质细胞、神经元和小胶质细胞中产生,是一个内源性抗炎性反应和神经保护因子[6]。吸入非常低浓度的H2S 都会产生强烈的刺激,所以直接吸入H2S 的安全性不清楚[7],试验中更多的用NaHS来研究H2S 的效应。在一个体外氧葡萄糖剥夺(oxygen glucose deprivation,OGD)模型,NaHS通过激活KATP/PKC/ERK1/HSP90路径减少了OGD之后的神经元损伤[8]。在一个体内卒中模型,发现NaHS保护海马神经元和改善了学习记忆障碍[9]。在帕金森病小鼠模型中发现吸入H2S有神经保护作用[10]。Ren 等[11]在15 min全脑缺血的动物模型中研究发现,H2S 对全脑缺血的预处理保护作用是剂量依赖性的,用低剂量的NaHS(25 μmol/kg)预处理,在全脑缺血后7 d评估,发现神经元损伤显著减轻,用高剂量的NaHS(180 μmol/kg)预处理却加重了神经元损伤,这与我们的研究结果相似,我们发现,相比较tGCI组,给予24 μmol/kg 的外源性NaHS后处理显著增加了全脑缺血大鼠海马神经元的存活细胞数目,但高剂量的 NaHS(180 μmol/kg)后处理没有提供保护效应。
Kv1.4和Kv4.2广泛分布于神经系统与心肌组织中,是神经元兴奋性的关键调节者,钾通道的改变与心脑缺血密切相关。Kv1.4和Kv4.2属于A型钾通道,其编码的是快速失活的瞬间外向钾电流IA,主要分布于可兴奋细胞中,参与调控动作电位的频率和激活阈值、动作电位的形态、递质释放和突触后兴奋性[12]。Kv1.4主要分布于神经细胞的轴突和末梢。Kv4.2分布在胞体和树突[13]。我们发现tGCI后海马区 Kv4.2和 Kv1.4 mRNA表达水平下调,这与许多类似研究结果一致。Liu等[14]在一个急性心肌梗死大鼠模型发现,心肌组织KV4.2的蛋白表达水平和Kv4.2组成的Ito电流的密度显著降低,并促成了急性心肌梗死后的心律失常。Lei 等[15]在大鼠短暂全脑缺血25 min之后2 m,观察到不论是在癫痫发作的缺血脑还是未发作癫痫的脑中,Kv4.2的表达水平都显著减少,其促成了缺血之后的癫痫发作及神经元损伤。另一方面,敲除Kv1.4和Kv4.2亚单位的转基因鼠培育的神经元展现出对缺血损伤的易损性增加[16]。当Kv4.2亚单位在遗传学上被删除时,海马CA1锥体细胞树突的IA电流几乎完全消失,LTP的阈值降低,并导致了海马功能的损伤。在体外和体内试验中均已证实,通过Kv1.4和Kv4.2重组体的共表达有效改善了不耐受缺血的中等大小的棘状神经元(medium spiny neurons,MS)的存活率[17]。我们发现与假手术组相比,各缺血再灌注组的Kv4.2和Kv1.4的mRNA表达水平均明显下调,甚至是在缺血再灌注之后48 h。Kv1.4和Kv4.2是A型钾通道的亚单位,介导了皮质和海马树突锥体神经元的兴奋性,其表达下调可能影响缺血后膜电位的稳定性,导致神经元对缺血的不耐受。
兴奋性中毒是许多神经系统疾病包括卒中的主要原因。IA电流调节神经元的兴奋性并因此影响病理学过程。我们推测Kv4.2的下调可能涉及脑缺血之后兴奋性中毒细胞的死亡。长久以来,人们认为钾电流的增强导致膜电位超极化并降低了兴奋性,对缺血是有保护作用的[16]。研究表明[18],在缺血损伤之后IA电流在纹状体的缺血耐受神经元中显著增强,IA的活化使峰阀值(spike threshold) 提高,缩短动作电位时程,从而降低神经元的兴奋性[19]。我们的研究发现,NaHS后处理显著增加了tGCI大鼠海马神经元的存活细胞数目,同时在再灌注26 h,与tGCI组比较,NaHS组Kv1.4和Kv4.2的mRNA表达水平均明显上调,暗示NaHS可能通过上调Kv1.4 和 Kv4.2 的 mRNA 表达发挥脑保护作用。其机制可能是:(1)根据Zou等人的研究,钾通道基因表达水平的变化与脑缺血时钾电流发生的变化相一致。在tGCI模型中,NaHS后处理使Kv4.2和K v1.4基因表达水平上调,增强了A-型钾电流,减慢了神经冲动的发放频率,缩短动作电位时程,抑制神经元兴奋性及降低氧耗,保护了神经元免受脑缺血损伤。(2)有研究显示[8],NaHS经由PKC/ERK1/2路径针对脑缺氧诱导的神经元死亡发挥了保护效应,而ERK/MAPK又通过对 Kv4.2的T607位点的直接磷酸化来动态调节通道功能[20]。那么NaHS也可能借由这些分子路径调节Kv4.2的基因及蛋白表达水平而发挥保护效应。在再灌注48 h,NaHS组与tGCI组相比较,Kv4.2和Kv1.4的mRNA表达差异无统计学意义,可能与实验中NaHS系单次给药,给药剂量偏小,导致其作用不持久有关。与KV4.2的表达不同的是,与假手术组相比,在再灌注26 h,NaHS组KV1.4 mRNA的表达水平较缺血组升高,但并未高于假手术组,这表明NaHS衰减了缺血之后kv1.4的降低,从而通过抑制IA电流衰减而发挥了脑保护作用。
总的来讲,我们的研究揭示了H2S 的潜在保护效应与其在脑卒中治疗中的潜在价值。
[1]Ruan YW,Ling GY,Zhang JL,et al. Apoptosis in the adult striatum after transient forebrain ischemia and the effects of ischemic severity[J]. Brain Res,2003,982(2):228-240.
[2]De Keyser J,Sulter G,Luiten PG. Clinical trials with neuroprotective drugs in acute ischaemic stroke:are we doing the right thing[J]. Trends Neurosci,1999,22(12):535-540.
[3]Huang H,Gao TM,Gong L,et al. Potassium channel blocker TEA prevents CA 1 hippocampal injury following transient forebrain ischemia in adult rats[J]. Neurosci Lett,2001,305:83-86.
[4]张海霞,李正斌,王晓良. 大脑中动脉栓塞模型大鼠的中枢电压依赖性钾通道 mR NA 表达的改变[J]. 药学学报,2006,41(4):328-332.
[5]Qu K. Hydrogen sulfide:neurochemistry and neurobiology[J]. Neurochem,2008,52:155-165.
[6]Lee M. Astrocytes produce the antiinflammatory and neuroprotective agent hydrogen sulfide[J]. Neurobiol,2009,30:1523-1534.
[7]Reiffenstein RJ. Toxicology of hydrogen sulfide[J]. Annu Rev Pharmacol Toxicol,1992,32:109-134.
[8]Tay AS. Hydrogen sulfide protects neurons against hypoxic injury via stimulation of ATP-sensitive potassium channel/protein kinase C/extracellular signal-regulated kinase/heat shock protein 90 pathway[J]. Neuroscience,2010,167:277-286.
[9]Li Z. Protective effects of exogenous hydrogen sulfide on neurons of hippocampus in a rat model of brain ischemia[J]. Neurochem Res,2011,36:1840-1849.
[10]Kida K. Inhaled hydrogen sulfide prevents neurodegeneration and movement disorder in a mouse model of Parkinson’s disease[J]. Antioxid Redox Signal,2011,15:343-352.
[11]Ren C,Du A,Li D,et al. Dynamic change of hydrogen sulfide during global cerebral ischemia-reperfusion and its effect in rats[J]. Brain Research,2010,1345(2):197-205.
[12]张亚男,马津全,王 舒. 钾通道与缺血性脑损伤的研究进展[J]. 中国脑血管病杂志,2012,9(4):217-220.
[13]黄 燕,王增贤. 膜片钳技术及其在星形胶质细胞钾离子通道研究中的应用[J]. 泰山医学院学报,2007,28(8):673-676.
[14]Liu X,Zhang Y,Du W,et al. MiR-223-3p as a Novel MicroRNA Regulator of Expression of Voltage-Gated K+ Channel Kv4.2 in Acute Myocardial Infarction[J]. Cell Physiol Biochem,2016,39(1):102-114.
[15]Lei Z,Zhang H,Liang Y. Reduced expression of IA channels is associated with post-ischemic seizures[J]. Epilepsy Res,2016,124:40-48.
[16]Deng P,Pang ZP,Lei Z. Up-regulation of A-type potassium currents protects neurons against cerebral ischemia[J]. J Cereb Blood Flow Metab,2011,31 (9):1823-35.
[17]Smith GD,Gao N,Lugo JN. Kv4.2 knockout mice display learning and memory deficits in the lashley maze[J]. F1000 Res,2016,5:1811-1132.
[18]Hu H,Shao LR,Chavoshy S,et al. Presynaptic Ca2+-activated K+channels in glutamatergic hippocampal terminals and their role in spike repolarization and regulation of transmitter release[J]. J Neurosci,2001,21(24):9585-9597.
[19]Pisani A,Bonsi P,Centonze D,et al. Involvement of intracellular calcium stores during oxygen/glucose deprivation in striatal large aspiny interneurons[J]. J Cereb Blood Flow Metab,2000,20(5):839-846.
[20]Laura A,Schrader,Shari G,et al. ERK/MAPK regulates the Kv4.2 potassium channel by direct phosphorylation of the pore-forming subunit[J]. Am J Physiol Cell Physiol,2006,290:C852-C861.
Effect of potassium channels Kv4.2 and Kv1.4 in neuroprotection of NaHS Postconditioning against Transient Global Cerebral Ischemia in Rats
BAIChengping,ZHAOChengliang.
(TheqinghaiuniversityaffiliatedhospitalneurologyDepartment,Xining810001,China)
Objective We intend to study the effects of sodium hydrosulfide postconditioning on the Kv4.2 and Kv1.4 potassium channel mRNA expression of hippocampus after transient global cerebral ischemia in rats and its role in brain protection,so as to explore the neuroprotective mechanism of NaHS against Transient Global Cerebral Ischemia model in Adult Rats. Methods Transient global ischemia was produced in adult Wistar rats using the 4-vessel occlusion method. The rats were randomly assigned into 3 groups:sham group,tGCI group,and tGCI+NaHS groups. NaHS postconditioning was carried out by intraperitoneal injection of 24 μmol/kg or 180 μmol/kg of NaHS at 1d post-tGCI. The neuronal death in the hippocampal CA1subregion was determined by Nissl staining and NeuN staining. The mRNA expression levels of Kv4.2 and Kv1.4 potassium channels were analyzed by RT-PCR. Results (1)Compared with the tGCI group,24 μmol/kg NaHS Postconditioning significantly increased the number of surviving cells in hippocampus. However,higher concentration of exogenous NaHS (180 μmol/kg) didn’t offer a protection against the neuronal injury induced by global cerebral ischemia-reperfusion. (2)The mRNA expression of Kv4.2 and Kv1.4 in hippocampus in the tGCI group were lower than that in Sham group (P<0.05). The mRNA expression of KV4.2 (1.24±0.08)、Kv1.4 (1.11±0.07) in hippocampus of NaHS treatment group was markedly higher than the tGCI group at 26 hours after reperfusion(0.75±0.04)、(0.79±0.06) respectively. (P<0.05). Conclusion Exogenous NaHS postconditioning may lead to membrane potential hyperpolarization by increasing Kv4.2 and Kv1.4 mRNA expression level,which inhibit the excitability of neurons and reduce oxygen consumption,protect neurons against cerebral ischemic injury.
Transient global cerebral ischemia; Potassium channels; NaHS neuroprotection
1003-2754(2017)07-0613-05
2017-03-23;
2017-06-10
青海省科技厅应用基础研究计划(No. 2016-ZJ-735)
(1.青海大学附属医院神经内科,青海 西宁 810001;2.青海大学研究生院,青海 西宁 810000) 通讯作者:拜承萍,E-mail:0101kobe@163.com
R743.31
A
猜你喜欢
浙江大学学报(医学版)(2023年6期)2023-05-11 11:09:30
今日农业(2020年20期)2020-12-15 15:53:19
能源(2018年10期)2018-12-08 08:02:48
中西医结合心血管病电子杂志(2018年28期)2018-11-19 11:04:54
能源(2016年10期)2016-02-28 11:33:30
中国康复理论与实践(2015年10期)2015-12-24 05:42:43
中国现代医学杂志(2015年26期)2015-12-23 11:04:20
吉林大学学报(医学版)(2015年5期)2015-12-16 15:43:56
中国体外循环杂志(2015年3期)2015-12-08 05:13:01
江苏农业科学(2015年1期)2015-04-17 23:53:02
