
高灵敏甲胎蛋白夹心免疫传感器的制备
2017-08-14 22:18:12王雪李鹏君邱萍王小磊
分析化学 2017年8期
王雪+李鹏君+邱萍+王小磊


摘 要 构建了新型甲胎蛋白(AFP)夹心免疫传感器。采用金纳米粒子-氧化石墨烯-普鲁士蓝纳米立方体(AuNP-GO-PBNCs)纳米复合材料标记甲胎蛋白(AFP)二抗,将制備的金-聚多巴胺-四氧化三铁(Au-PDA-Fe3O4)磁性纳米复合物固定在自制的磁性电极表面,通过吸附作用固定AFP一抗,用牛血清白蛋白(BSA)封闭电极上的非特异性吸附位点。在37℃下与AFP抗原溶液孵育50 min,最后将电极放入AuNP-GO-PBNCs纳米复合材料标记的二抗溶液中孵育,基于此建立了采用普鲁士蓝(PB)标记的的夹心免疫传感器检测AFP的方法。在最佳实验条件下,PB催化H2O2氧化的响应电流与AFP的浓度表现出两段线性关系,线性范围分别为0.005~1.000 ng/mL和1~20 ng/mL, 检出限(LOD, S/N=3)为1.0 pg/mL。本方法具有灵敏度高、选择性好的特点。
关键词 金纳米粒子; 氧化石墨烯; 甲胎蛋白; 普鲁士蓝; 夹心免疫传感器
1 引 言
甲胎蛋白(AFP)是临床诊断中常用的特异性的肿瘤标志物,对原发性肝癌的早期诊断具有重要的临床意义。目前,检测血清中甲胎蛋白主要采用放射免疫法、电化学发光法、化学发光法和酶联免疫法等[1~5]。放射免疫法检测AFP能达到纳克水平,但存在放射性污染; 电化学发光法检测速度快,但检测成本比酶联免疫法高; 化学发光法技术较为成熟,但是检测时间较长; 酶联免疫法(ELISA)具有简单、灵敏、特异、快速及易于自动化操作等特点,在生物工程[6]、临床诊断[7~9]、药物及环境分析[10~12]等方面应用广泛。ELISA法通常采用酶标记抗体(或者抗原),标记酶通常是分子量小、易制备的辣根过氧化酶(HRP)[7,12~14]。然而,HRP的本质是蛋白质,使用过程中存在易失活的问题。
为了克服HRP等标记酶易失活的缺点,很多具有优良类过氧化酶催化活性的纳米材料被应用于ELISA检测方法中[8~10],其中普鲁士蓝(PB)具有独特的三维网状结构,具备稳定性好且制备成本低的优点。另外,它还具有优良的电化学可逆性,作为电子转移媒介体,对过氧化氢表现出良好的电催化能力[15]。使用PB代替常规酶蛋白标记抗体或抗原,可以克服现有酶标记方法操作繁琐、成本高、酶容易失活等缺陷。石墨烯具有导电性强、比表面积大、成本低等优点[16~19]。将石墨烯做为PB的载体,可大大提高PB的负载量; 此外,它可以增加普鲁士蓝的稳定性[20]。Fe3O4磁性纳米材料具有良好的超顺磁性、低毒性和生物兼容性,在生物催化、生物标记等领域得到广泛应用[21~24]。磁性纳米材料具有大的表面Gibbs自由能,可以降低体系的能量,化学稳定性高。
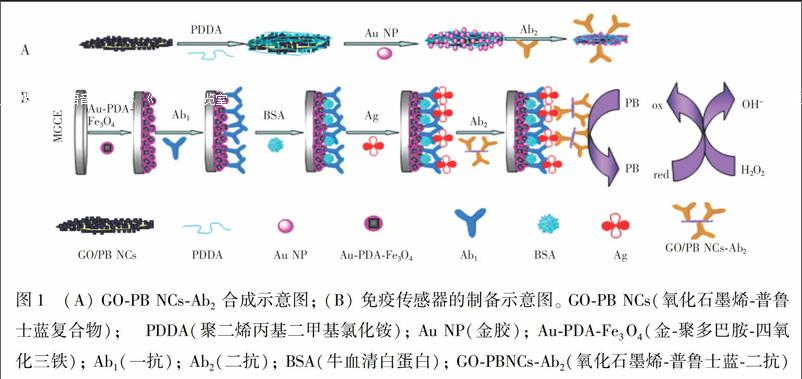
本研究制备了氧化石墨烯/普鲁士蓝(GO-PBNCs)复合物,用聚二烯丙基二甲基氯化铵(PDDA)功能化的带正电的GO-PBNCs吸附带负电的金纳米粒子(AuNPs),制得AuNP-GO-PBNCs纳米复合材料,用此纳米复合材料标记AFP二抗(Ab2)。另外合成了磁性Fe3O4立方体,基于多巴胺(DA)与HAuCl4·4H2O反应可生成聚多巴胺(PDA) 及金纳米粒子,采用原位化学氧化聚合的方法制得Au-PDA-Fe3O4磁性纳米复合物。利用磁性将Au-PDA-Fe3O4磁性纳米复合物固定在自制的磁性电极表面后[25],吸附固定anti-AFP,制备检测AFP的磁性传感电极。将此传感电极与含有AFP抗原的样品孵育; 再将已结合AFP抗原的电极与AuNP-GO-PBNCs纳米复合材料标记的二抗孵育,测定电极的电化学响应信号。制备的以普鲁士蓝为标记物的夹心免疫传感器具有灵敏度高、检出限低和稳定性好的特性,在生物医学、临床诊断等方面有潜在的应用价值。
2 实验部分
2.1 仪器与试剂
Quanta 200扫描电子显微镜(SEM,,美国FEI公司); UV-2450紫外可见分光光度计(日本Shimadzu公司); X-射线衍射仪 (XRD, 英国Bede公司); Autolab PGSTAT30/FRA2 电化学工作站(瑞士Metrohm公司)。采用三电极工作系统,修饰的磁性玻碳电极(MGCE)作为工作电极,饱和甘汞电极(SCE) 作为参比电极,铂丝电极作为对电极(CE)。
甲胎蛋白(AFP)、anti-AFP(博赛生物有限公司); 天然石墨片(99.8%,325目)、多巴胺(DA,99%)(Alfa Aesar公司); 氯金酸(HAuCl4·4H2O,99.8% )、牛血清白蛋白(BSA,≥98%) (Sigma公司)。以0.1 mol/L Na2HPO4-NaH2PO4磷酸盐缓冲液(PBS, pH 5.91)为支持电解质溶液。其它试剂均为国产分析纯,使用前未经进一步处理。实验用水为超纯水(≥18 MΩ cm)。
2.2 实验方法2.2.1 Au-PDA-Fe3O4磁性纳米复合材料的制备
根据文献[26]制备磁性Fe3O4纳米立方体。将Fe3O4纳米立方体加入到1.0 mL PBS缓冲溶液中,浓度为0.5 mg/mL,在4℃剧烈搅拌条件下,加入500 μL 60 mmol/L (过量)多巴胺,再逐滴加入300 μL 2%(m/V) HAuCl4溶液,继续搅拌30 min,得到Au-PDA-Fe3O4磁性纳米复合材料。
2.2.2 Ab2-AuNP-GO-PBNCs生物复合材料的制备 采用Hummers方法合成氧化石墨烯(GO) [27,28]。在超声条件下,用水将GO稀释成1 mg/mL的悬浮液。取5 mL加入圆底烧瓶中,再加入0.2 mL 5 mmol/L FeCl3-K3[Fe(CN)6](pH 1.6),置于30℃水浴中搅拌反应2 h,溶液颜色由黄棕色变为深蓝绿色,即得GO-PBNCs复合物。离心,用水清洗沉淀,再离心,至上清液为无色。如图 1A 所示,在超声条件下,将制得的GO-PBNCs复合材料加入到含0.625 mol/L KCl和1.25 mg/mL PDDA溶液中,继续超声处理3 h,离心清洗后,得到PDDA@GO-PB NCs复合材料,然后将其分散到5 mL 水中。根据文献[29]方法制得AuNPs,在超声条件下向10 mL AuNPs溶液中加入0.5 mL制得的PDDA@GO-PB NCs复合材料,放置过夜,次日离心,清洗后,将其分散在1 mL PBS缓冲溶液中,加入150 μL 2.0 μg/mL Ab2,在4℃下反应12 h后,离心,用100 μL 1%(w/w) BSA封闭,得到Ab2-AuNP-GO-PBNCs复合材料。离心,沉淀分散在1 mL PBS溶液中,待用。
2.2.3 制备修饰电极及检测AFP 依据文献[30]制备磁性玻碳电极(MGCE)。将10 mg石墨粉和1.2 mL石蜡油混合均匀,调成碳糊,然后将适量碳糊填入长50 mm、直徑3 mm聚四氟乙烯管中,将铜丝插在聚四氟乙烯管的一端,将一个厚2 mm、直径3 mm的圆形磁铁(其表面可产生0.2 T的非均匀磁场)放在距离管口约2 mm处的另一端,再在管口处放进一个厚2 mm、直径3 mm的玻碳,将磁铁与玻碳紧紧连在一起,并将玻碳的边沿用胶密封起来,即得到磁性玻碳电极。
将制备好的MGCE依次用1.0、0.3 和0.05 μm的Al2O3在麂皮上抛光至镜面后,再依次用超纯水、无水乙醇、0.1 mol/L HNO3、超纯水超声清洗,氮气吹干。吸取6 μL Au-PDA-Fe3O4磁性纳米复合材料悬浮液,小心滴涂在MGCE表面,在磁场的作用下,电极表面紧紧固定一层Au-PDA-Fe3O4磁性纳米复合材料,室温下自然干燥,制得Au-PDA-Fe3O4修饰的MGCE。将已修饰好的MGCE浸入AFP一抗溶液中,在4℃条件下过夜。在室温下,将电极浸入1% BSA溶液中约1 h,封闭电极上的非特异性吸附位点,即制得免疫传感器。将制备好的传感器分别浸入到不同浓度的AFP溶液中孵育50 min,吸取10 μL Ab2-AuNP-GO-PBNCs溶液,小心滴涂到修饰好的电极上,放置约1 h后,再用PBS缓冲液漂洗。将修饰好的电极浸入在含有1 mmol/L H2O2的PBS溶液(pH 5.91)中,采用差分脉冲伏安法(DPV)进行检测,免疫传感器的制备以及检测过程如图1B所示。
3 结果与讨论
3.1 GO-PBNCs与AuNP-GO-PBNCs复合材料的表征
由GO-PBNCs的扫描电镜图(图2A)可见,大小均一的PBNCs附着在片状的石墨烯表面; 用带正电的PDDA功能化并吸附带负电的AuNPs,其表面附着了一层均匀的Au NPs(图2B)。UV-vis表征结果如图2C所示,曲线a是GO的吸收光谱, 230 nm处的吸收对应C=C双键的π-π*跃迁[29]; 曲线b是GO-PBNCs复合物的吸收光谱,由于催化还原Fe3+,GO表面的电子密度发生了转移,所以在230 nm处没有明显的吸收。而由于PB中的Fe2+与Fe3+的电荷转移[28],GO-PBNCs复合物约在740 nm处出现了一个宽峰,说明在GO上成功生成PB[31]。曲线c在530 nm处有一吸收峰,为AuNPs的特征峰[32],这是由于用PDDA功能化后的GO-PBNCs复合物带正电,并吸附了带负电的AuNPs后,这进一步证实已成功合成AuNP-GO-PBNCs复合材料,上述结果与电镜表征结果相一致。
3.2 Fe3O4与Au-PDA-Fe3O4磁性纳米复合材料的表征
扫描电子显微镜(SEM)表征结果(图3A)显示,制备的Fe3O4的平均粒径为30 nm,颗粒大小均匀。图3B是Au-PDA-Fe3O4复合纳米粒子的SEM图像,未观察到Fe3O4立方体粒子,这可能是由于利用多巴胺原位合成金纳米粒子时, Fe3O4纳米立方体被包裹在内部。图3C是Fe3O4纳米立方体和Au-PDA-Fe3O4的UV-Vis图谱,Fe3O4纳米立方体没有明显的紫外吸收(曲线a); 曲线b是PDA的紫外吸收图谱,在280 nm出现了多巴胺的特征吸收[33]; 曲线c在530 nm处出现了金纳米颗粒的特征吸收峰,表明在Fe3O4纳米立方体表面确实形成了金纳米层[34],与电镜的表征结果(图3B)一致。
图4为Fe3O4纳米立方体和Au-PDA-Fe3O4的XRD图谱。在图4a上可见2θ=30.2°(220)、35.5°(311)、43.2°(400)、53.6°(422)、57.08°(511)和62.9°(440)等处的衍射峰,与ASTM标准数据卡片中Fe3O4图相吻合,表明合成的物质是立方尖晶石结构的Fe3O4[35]。图4b显示,Au-PDA-Fe3O4复合物产物在2θ=38.1°、44.4°、64.6°和77.6°等处出现了清晰的特征衍射峰,分别对应于Au (111)、(200)、(220)和(311)晶面,表明产物表面已经覆盖了一层金纳米粒子[36]。另外,因为Au是重金属元素,且前文提到Fe3O4的外表面覆盖了一层金,而Fe3O4的衍射峰相对Au的衍射峰太弱,因此很难观测到。插图是在外磁场条件下拍摄的,Fe3O4悬浮液呈黑色,且均匀分散,若将一块磁铁放置在该悬浮液的一侧,施加磁场,所有的Fe3O4立即聚沉,上清液则澄清透明。以上结果充分表明,Fe3O4纳米立方体具有良好的超顺磁性,在外加磁场的作用下,可以将其固定在磁性玻碳电极(MGCE)表面[37,38]。
3.3 修饰电极的电化学行为
图5A是在含有探针分子[Fe(CN)6]3/4 (5.0 mmol/L)的电解质溶液中,不同的修饰电极的交流阻抗复平面图。曲线a为裸电极阻抗图,在所有频率范围内几乎呈一条直线,表明探针分子非常容易到达MGCE电极表面发生氧化还原反应,这一电化学过程是受扩散控制的。当在MGCE电极表面固定Fe3O4-PDA-Au时, Fe3O4-PDA-Au阻碍了探针分子向电极表面的扩散,曲线在高频区出现一个显著的半圆,半圆直径对应于电极的电子传递阻抗(Rct),约145 Ω(曲线b)。Fe3O4-PDA-Au修饰电极吸附anti-AFP后(曲线c), Rct增大,表明anti-AFP已成功吸附在电极MGCE/Fe3O4-PDA-Au上。用BSA封闭MGCE/Fe3O4-PDA-Au/anti-AFP 上的非特异性吸附位点即得MGCE/Fe3O4-PDA-Au/anti-AFP/BSA (曲线d),再与AFP孵育发生免疫反应后得到MGCE/Fe3O4-PDA-Au/anti-AFP/BSA /AFP (曲线e),Rct逐渐增大,这证明AFP与anti-AFP发生了免疫反应后,成功组装在A修饰电极表面上,也证明了Fe3O4-PDA-Au可以保持anti-AFP的生物活性。然而,当MGCE/Fe3O4-PDA-Au/anti-AFP/BSA /AFP进一步与GO-PB-Au NPs-Ab2孵育后(曲线f),Rct相对发生免疫反应后减小,这是由于GO和Au NPs具有很好的导电性,从而使得探针分子能够较容易的到达电极表面。
電压在0.2~0.5 V范围内,扫描速度为50 mV/s,对不同修饰电极进行循环伏安扫描(图5B)。PDA-Au/anti-AFP/BSA/AFP修饰电极未出现明显的循环伏安峰,当电极修饰Fe3O4-PDA-Au/anti-AFP/BSA/AFP后,背景电流显著增大,说明Fe3O4具有很强的导电性,起到增加电流的响应信号的作用; 当进一步孵育GO-PB-Au NPs-Ab2后,出现了一对对称的氧化还原峰,这是因为PB是很好的氧化还原活性分子,当在支持电解质溶液中加入1 mmol/L H2O2后,还原峰电流明显增大。因此,PB不仅起到了电子介体的作用,而且可作为一种“人工过氧化酶”,起到了电催化的作用。
3.4 孵化时间和温度的影响
对免疫孵育时间进行了优化。随着孵育时间的延长,响应电流也随之增大,当孵育时间超过50 min后,响应电流信号基本保持不变,这表明当孵育时间大于50 min时,电极上结合的AFP达到饱和,因此选择50 min为最佳孵育时间。
温度也是影响免疫反应和抗原抗体稳定性的重要因素。在15~50℃范围内,免疫传感器的电流响应信号随着温度的升高而先增大后降低,在37℃时达到最大,这是因为温度的升高有利于提高抗原与抗体的反应。而温度过高时,可能会导致抗原与抗体的蛋白分子变性,结合活性降低,使得组装上的GO-PB-AuNPs-Ab2减少,从而相应的PB及催化H2O2的响应电流减小。因此选择37℃为最佳测定温度。
3.5 免疫传感器的安培响应
在最佳实验条件下,采用DPV检测不同浓度AFP的结果如图6所示。在不同浓度的AFP存在下,传感器的电流响应信号表现出两段线性关系,线性范围分别为 0.005~1.000 ng/mL和1~20 ng/mL,检出限为1.0 pg/mL(S/N=3)。本方法与文献报道方法的分析性能的比较见表1。
3.6 免疫传感器的重现性和稳定性
用同一根免疫电极对5 ng/mL AFP标准溶液进行重复测定6次,测量电化学响应信号的RSD为2.7%。取6支同样条件下制备的的免疫电极,对5 ng/mL AFP样品进行检测,测定结果的RSD为4.3%,表明此传感器具有良好的重现性。将制备好的免疫传感器在4℃下放置30天后重新检测,测得的电流值是初始电流响应的93%,表明此传感器具有良好的长期稳定性。
3.7 免疫传感器的选择性
为了进一步研究传感器测定AFP的选择性,在最佳条件下,测定了免疫传感器对5 ng/mL AFP及其它干扰物包括癌胚抗原(CEA,5 ng/mL)、牛血清白蛋白(BSA,5 ng/mL)、乙肝表面抗原(HBsAg, 5 ng/mL)的响应,结果如图7所示,免疫传感器对AFP的响应远大于其它干扰物,表明制备的免疫传感器的抗干扰能力强,选择性好。
4 结 论
以MGCE作为电极,基于AuNP-GO-PBNCs复合材料成功构建了一种新型的甲胎蛋白夹心免疫传感器,实现了对AFP的灵敏检测。此传感器灵敏度高,稳定性好,线性范围宽且成本低。所制备的AuNP-GO-PBNCs复合材料具有良好的导电性和大的比表面积,又可以作为电子介体催化过氧化氢,起到放大信号的作用。此免疫传感器制作简单,使用方便,成本低,在生物医学、临床诊断、健康检测等方面具有潜在的应用价值。
References
1 Maeda M. J. Pharmaceut. Biomed., 2003, 30(6): 1725-1734
2 ZHANG Jun-Rui, CHEN Jian, LIU Zhong-Ming. Chinese J. Anal. Chem., 2010, 38(8): 1219-1226
张军瑞, 陈 健, 刘仲明. 分析化学, 2010, 38(8): 1219-1226
3 SHEN Hai-Ying, WANG Xiao-Qian, WANG Yun-Yun, QU Feng. Chinese J. Anal. Chem., 2017, 45(1): 83-88
沈海滢, 王晓倩, 王云云, 屈 峰. 分析化学, 2017, 45(1): 83-88
4 ZHU Yu-Ping, HE Wei, WANG Bi. Journal of Instrumental Analysis, 2013, 32(11): 1322-1327
朱宇萍, 何 伟, 王 碧. 分析测试学报, 2013, 32(11): 1322-1327
5 ZHOU Xue, OUYANG Wu-Qing, WEI Yun-Peng, SHEN Fang-Chao. Chinese J. Anal.Chem., 2015, 43(10): 1589-1593
周 雪, 欧阳五庆, 魏云鹏, 申方超. 分析化学, 2015, 43(10): 1589-1593
6 Chung C, Makino R, Kong J, Ueda H. Anal. Chem., 2015, 87(6): 3513-3519
7 Singh V, Krishnan S. Anal. Chem., 2015, 87(5): 2648-2654
8 Sun J, Hu T, Chen C, Zhao D, Yang F, Yang X. Anal. Chem., 2016, 88(19): 9789-9795
9 Sun J, Hu T, Xu X, Wang L, Yang X. Nanoscale, 2016, 8(38): 16846-16850
10 Wang S, Chen Z, Choo J, Chen L. Anal. Bioanal. Chem., 2016, 408(4): 1015-1022
11 Lotierzo M, Abuknesha R, Davis F, Tothill I E. Enviro. Sci. Technol., 2012, 46(10): 5504-5510
12 Hirakawa Y, Yamasaki T, Harada A, Ohtake T, Adachi K, Iwasa S, Narita H, Miyake S. J. Agric. Food Chem., 2015, 63(36): 8075-8082
13 Sathe M, Srivastava S, Agrawal S K. Defence Sci. J., 2016, 66(5): 471-478
14 Du D, Wang L, Shao Y, Wang J, Engelhard M H, Lin Y. Anal. Chem., 2011, 83(3): 746-752
15 Su L J, Xiong Y H, Yang H G. J. Mater. Chem. B, 2016, 4(1): 128-134
16 Talyzin A V, Luzan S M. J. Phys. Chem. C, 2010, 114(15): 7004-7006
17 Talyzin A V, Sundqvist B, Szabo T, Dekany I, Dmitriev V. J. Am. Chem. Soc., 2009, 131(51): 18445-18449
18 Jin C, Lee J, Lee E, Hwang E, Lee H. Chem. Commun., 2012, 48(35): 4235-4237
19 Sun J Y, Huang K J, Zhao S F, Fan Y, Wu Z W. Bioelectrochem., 2011, 82(2): 125-130
20 Qiu W, Zhu Q, Gao F, Gao F, Huang J, Pan Y, Wang Q. Mat. Sci. Eng. C, 2017, 72: 692-700
21 Li Z, Potapenko D, Rim K, Flytzani-Stephanopoulos M, Flynn G, Osgood R, Wen X, Batista E. J. Phys. Chem. C, 2015, 119(2): 1113-1120
22 Lin T, Wang J, Guo L, Fu F. J. Phys. Chem. C, 2015, 119(24): 13658-13664
23 Lee J, Kwon S, Park J, Hyeon T. Nano Lett., 2015, 15(7): 4337-4342
24 Lee N, Schuck P, Nico P, Gilbert B. J. Phys.Chem. Lett., 2015, 6(6): 970-974
25 Huang Z, Han W, Wu Y, Hu X, Yuan Y, Chen W, Peng H, Liu A, Lin X. J. Electronal. Chem., 2017, 785: 8-13
26 Goon I Y, Zhang C, Lim M, Gooding J J, Amal R. Langmuir, 2010, 26(14): 12247-12252
27 Hummers W S, Offeman R E. J. Am. Chem. Soc., 1958, 80(6): 1339-1339
28 Tang L, Wang Y, Li Y, Feng H, Lu J, Li J. Adv. Funct. Mater., 2009, 19(17): 2782-2789
29 He Y, Zhang X, Zhang S, Kris M, Man F C, Kawde A. Biosens. Bioelectron., 2012, 34(1): 37-43
30 Wang J, Kawde A N. Electrochem. Commun., 2002, 4(4): 349-352
31 Chen H, Ma Y, Wang X, Wu X, Zha Z. RSC Adv., 2017, 7(1) : 248-255
32 Wu J, Xiang D, Reuven Gordon. Anal. Chem., 2017, 89(4): 2196-2200
33 Chen J L, Yan X P, Meng K, Wang S F. Anal. Chem., 2011, 83(22): 8787-8793
34 Vaucher S, Li M, Mann S. Angew. Chem. Int. Ed., 2000, 39(10): 1793-1796
35 Liang R P, Meng X Y, Liu C M, Qiu J D. Electrophoresis, 2011, 32(23): 3331-3340
36 Si J Y, Yang H. Mater. Chem. Phys., 2011, 28(3): 519-524
37 Peng H, Liang R, Zhang L, Qiu J. J. Electronal. Chem., 2013, 700: 70-76
38 Zou C, Fun Y, Xie Q, Yao S. Biosens. Bioelectron., 2010, 25(6): 1277-1282
39 Niu Y L, Yang T, Ma S S, Peng F, Yi M H, Wan M M, Mao C, Shen J. Biosens. Bioelectron., 2017, 92: 1-7
40 Liu Y C, Chen J Y, Du M Y, Wang X X, Ji X H, He Z K. Biosens. Bioelectron., 2017, 92: 68-73
猜你喜欢
肝博士(2024年5期)2024-01-01 00:00:00
保健医苑(2022年10期)2022-10-29 04:37:44
中老年保健(2019年10期)2019-10-19 09:30:08
现代商贸工业(2016年31期)2017-04-06 15:26:12
纺织导报(2016年12期)2017-01-06 12:11:11
科技视界(2016年12期)2016-05-25 20:00:06
中国科技博览(2016年9期)2016-04-25 10:22:52
肝博士(2015年3期)2015-09-25 09:19:44
湖南大学学报·自然科学版(2015年6期)2015-07-20 18:21:35
中国现代医生(2014年17期)2014-07-05 01:02:26
