
离子色谱法测定水果和蔬菜中3-吲哚乙酸和3-吲哚丁酸的含量
2017-07-20 10:09赵振东朱建忠
理化检验-化学分册 2017年5期
赵振东,李 平,朱建忠
(1.海南大学材料与化工学院,海口570228; 2.海南国际旅游岛发展研究院,海口570228;3.海南赛诺实业有限公司,海口570125)
离子色谱法测定水果和蔬菜中3-吲哚乙酸和3-吲哚丁酸的含量
赵振东1,2,李 平3,朱建忠1
(1.海南大学材料与化工学院,海口570228; 2.海南国际旅游岛发展研究院,海口570228;3.海南赛诺实业有限公司,海口570125)
水果或蔬菜样品(5.00g)采用含10%(体积分数)乙腈的35mmol·L-1氢氧化钠溶液45mL超声提取,萃取液经固相萃取C18小柱净化后,选用AS11分离柱(4mm×250mm)进行色谱分离,用与上文中相同的乙腈-氢氧化钠混合液为流动相。用紫外检测器测定。3-吲哚乙酸和3-吲哚丁酸的质量浓度均在0.05~10g·L-1内与其对应的峰面积呈线性关系,两者的检出限(3S/N)依次为0.009,0.012g·L-1。对10g·L-1的3-吲哚乙酸和3-吲哚丁酸的混合标准溶液连续测定6次,峰面积的相对标准偏差依次为0.14%,1.4%。在0.50,2.0,10g·L-1等3个浓度水平进行加标回收试验,回收率在77.1%~104%之间。
离子色谱法;3-吲哚乙酸;3-吲哚丁酸;水果;蔬菜
3-吲哚乙酸和3-吲哚丁酸具有植物生长调节作用,广泛应用于农业生长调节和水果蔬菜催熟等方面。由于不规范使用,其对当前环境的影响日趋严重,引起了人们极大的关注。目前对3-吲哚乙酸和3-吲哚丁酸的测定方法主要有气相色谱法、放射免疫法、酶联免疫法和高效液相色谱法等[16]。这些方法存在样品处理操作步骤繁琐,所测成分中途损失严重,获得的数据可靠性较低等缺点[7]。也有使用离子色谱-电导检测和离子色谱-荧光检测的相关报道[8],但尚未见离子色谱-紫外检测的方法。本工作建立了离子色谱-紫外检测对3-吲哚乙酸和3-吲哚丁酸的残留量的分析方法。
1 试验部分
1.1 仪器与试剂
CIC 500型离子色谱仪,配二元泵、柱温箱、淋洗液发生器、自动进样器和紫外检测器;LC 30AD型液相色谱仪;JYL-C50T型粉碎机;SB-3200DTS型超声波萃取仪;PureLab Ultra超纯水系统;离子色谱专用固相萃取(SPE)C18小柱(300mg)。
3-吲哚乙酸和3-吲哚丁酸标准储备溶液:1.000g·L-1,称取3-吲哚乙酸标准品(纯度为99.5%)和3-吲哚丁酸标准品(纯度为99.9%)各10.05,10.01mg置于10mL容量瓶中,用流动相溶解(可超声波助溶解)。

图1 色谱图Fig.1 Chromatograms
乙腈为色谱纯,试验用水为超纯水。
1.2 色谱条件
AG11保护柱(4mm×50mm),AS11分离柱(4mm×250mm);柱温30℃;流动相为含10%(体积分数,下同)乙腈的35mmol·L-1氢氧化钠溶液,流量1.0 mL·min-1;进样体积25μL,分析时间30min;紫外检测波长210nm。
1.3 试验方法
称取经粉碎机打粉后的试样5.00g于50mL容量瓶中,加入流动相45mL作为提取溶液,密封超声萃取30min,静置至室温后加入流动相定容,转移部分溶液至离心管中,于4℃下,以10 000r· min-1转速离心20min。上清液经0.45μm有机滤膜过滤,滤液经SPE C18小柱净化后,按仪器工作条件进行测定。
2 结果与讨论
2.1 色谱行为
3-吲哚乙酸和3-吲哚丁酸标准溶液、豇豆空白样品和豇豆加标样品的色谱图见图1。
由图1可知:3-吲哚乙酸和3-吲哚丁酸的保留时间分别为2.8,4.8min。
2.2 样品提取方法的选择
水果和蔬菜中农药残留的提取方法主要采用水浴摇床萃取、加速溶剂萃取和匀浆萃取等,这些方法操作过程稍繁琐,提取效率欠佳,同时分析成本高。超声萃取具有提取时间短、效率高和分析成本低的特点。试验选用超声萃取法提取水果、蔬菜样品中农药残留。
3-吲哚乙酸和3-吲哚丁酸在强碱性溶剂中主要呈阴离子状态存在,由于具备π电子共轭体系,它们在水中的溶解度较小,故在提取溶剂中加入少量乙腈有助于提高渗透效率。
2.3 检测波长的选择
在不连接色谱柱的情况下,采用含10%乙腈的35mmol·L-1氢氧化钠溶液作为流动相,对10g· L-1的3-吲哚乙酸和3-吲哚丁酸混合标准溶液在配二极管阵列检测器的液相色谱仪中进行全扫描(190~600nm)。结果表明:3-吲哚乙酸和3-吲哚丁酸在波长210nm处有较强的紫外吸收。进一步对空白样品进行扫描发现,在波长210nm处干扰物质吸收不强,便于检测。试验选择检测波长为210nm。
2.4 色谱条件的选择
2.4.1 色谱柱
水果和蔬菜中化学成分比较复杂,化学性质差别明显,只有选择合适的色谱柱对目标组分进行分离,才能达到有效分离的目的。试验考察了AS11分离柱(4mm×250mm)和AS19分离柱(4mm× 250mm)对标准品和实际样品的分离效果。结果表明:AS11分离柱能有效将3-吲哚乙酸、3-吲哚丁酸与其他共存离子分离,峰形较好,分析时间短。此外,使用AS11分离,柱压较低,在连续分析复杂样品时,能起到保护泵的作用。试验选用AS11阴离子柱(4mm×250mm)为分离柱。
2.4.2 淋洗液
准确测定目标离子,必须选择合适的淋洗液,保证被测组分与其他离子能有效分离。目前用于分离阴离子的淋洗液有碳酸盐和氢氧根体系;其中氢氧根体系对于有机酸的分离最佳,且无需单独配制,工作效率比较高。试验选用氢氧根体系的淋洗液。
3-吲哚乙酸和3-吲哚丁酸由于具备π电子共轭体系,会与色谱柱填料中π电子产生共轭效应,导致长期保留在色谱柱内,因此试验在淋洗液中加入一定量的乙腈(乙腈的加入不影响紫外检测),利用乙腈屏蔽色谱柱中的π电子对。试验考察了在淋洗液中加入不同体积分数的乙腈对测定结果的影响。结果表明:乙腈的体积分数为15%时,色谱保留行为太弱,目标组分与干扰组分共流出;乙腈的体积分数为5%时,目标组分保留时间过长,同时重复性变差;乙腈的体积分数为10%时,既能保证目标组分与干扰离子的有效分离,又节约分析时间。试验选择在淋洗液中加入10%乙腈。
2.5 标准曲线和检出限
移取1 000g·L-1的3-吲哚乙酸和3-吲哚丁酸标准储备溶液,配成0.05,0.10,1.0,5.0,10g· L-1的3-吲哚乙酸和3-吲哚丁酸标准溶液系列,按色谱条件对其进行测定,以3-吲哚乙酸和3-吲哚丁酸的质量浓度为横坐标,对应的峰面积为纵坐标绘制标准曲线。结果表明:3-吲哚乙酸和3-吲哚丁酸的质量浓度均在0.05~10g·L-1内呈线性,线性回归方程和相关系数见表1。
取等比例空白基质样品,混合后加入已知量的标准溶液,按试验方法进行测定,采用逐级稀释方法,以3倍信噪比计算方法的检出限(3S/N),以10倍信噪比计算方法的测定下限(10S/N),结果见表1。

表1 线性回归方程、相关系数、检出限和测定下限Tab.1 Linear regression equations,correlation coefficients,detection limits and lower limits of determination
2.6 精密度试验
按试验方法对10g·L-1混合标准溶液连续测定6次,结果表明:3-吲哚乙酸和3-吲哚丁酸保留时间的相对标准偏差(RSD)分别为0.20%,1.2%;3-吲哚乙酸和3-吲哚丁酸峰面积的RSD分别为0.14%,1.4%。
2.7 回收试验
采用1 000g·L-1的3-吲哚乙酸和3-吲哚丁酸标准溶液,配成0.50,2.0,10g·L-1的提取液。称取打浆后的水果和蔬菜空白样品各5g,依次加入3种不同浓度加标提取液,按试验方法进行测定,每个平行样品3份,其结果见表2。
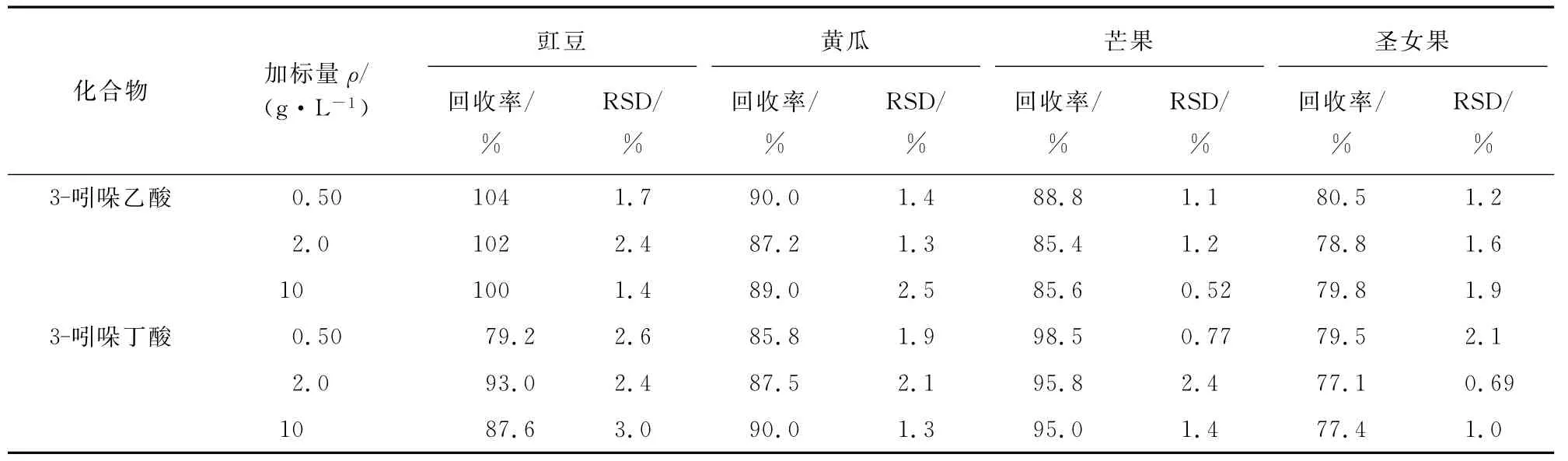
表2 回收试验结果(n=3)Tab.2 Results of test for recovery(n=3)
本工作采用离子色谱-紫外检测的方法快速测定水果和蔬菜中3-吲哚乙酸和3-吲哚丁酸的残留量,相比于其他方法,本法前处理更为简单、准确、快捷,灵敏度高,特别适用于大量样品检测。
[1] 李惠琳,云自厚,杨亦平,等.α-溴-2、3、4、5、6五氟甲苯衍生气相色谱同时测定吲-3-乙酸和脱落酸[J].植物学报,1990,32(3):205-209.
[2] ESPSTEIN E,COHEN J D.Microscale preparation of pentafluorobenzyl esters:dlectron-capture gas chromatographic detection of indole-3-acetic acid from plants[J].Journal of Chromatography,1981,209(3):413-420.
[3] 吴少伯,黄海,赵毓桔.植物激素脱落酸的放射免疫测定[J].植物生物学通讯,1985(1):44-46.
[4] 吴颂如,陈婉芬,周夔.酶联免疫法(ELISA)测定内源植物激素[J].植物生物学通讯,1988(5):53-57.
[5] 谢君,张义正.植物内源激素的反相高效液相色谱法测定[J].分析测试学报,2001,20(1):60-61.
[6] 张培志,徐育,朱岩.离子色谱法分离-抑制电导检测吲哚乙酸和吲哚丁酸[J].理化检验-化学分册,2002,38(9):448-449.
[7] 吴东亮,徐茂生,张培志,等.离子色谱法电导检测测定土壤中的吲哚-3-乙酸[J].仪器仪表学报,2001,22(4):376-377.
[8] 施青红,吴东亮,陈志斌,等.离子色谱荧光检测植物生长调节素吲哚-3-乙酸和吲哚-3-丁酸[J].宁波高等专科学校学报,2001,13:147-150.
IC Determination of Indol-3-acetic Acid and Indol-3-butyric Acid in Fruit and Vegetable
ZHAO Zhendong1,2,LI Ping3,ZHU Jianzhong1
(1.College of Materials and Chemical Engineering,Hainan University,Haikou570228,China;2.Institute for the Development of Hainan International Tourism Island,Haikou570228,China;3.Hainan Shiner Industrial Co.,Ltd.,Haikou570125,China)
The sample of fruit or vegetable(5.00g)was extracted ultrasonically with(45mL)of a mixture of CH3CN and 35mmol·L-1NaOH solution(10+90),and the extract was then purified on C18SPE colunm.The purified extract was separated by IC,using AS11column(4 mm×250 mm)as stationary phase and the same mixture of CH3CN and NaOH solution mentioned above as mobile phase.UV detector was used in the determination.Linear relationships between values of mass concentration of indol-3-acetic acid and indol-3-butyric acid were kept in the same range of 0.05-10g·L-1,with detection limits(3S/N)of 0.009,0.012g·L-1respectively.Precision of the method was tested at the concentration level of 10g·L-1indol-3-acetic acid and indol-3-butyric acid mixed standard solution for 6determination,RSD′s found from the peak area values were 0.14%and 1.4%respectively.Tests for recovery were made by standard addition method at the concentration levels of 0.50,2.0,10g·L-1,giving values of recovery in the range of 77.1%-104%.
IC;indol-3-acetic acid;indol-3-butyric acid;fruit;vegetable
O657.7
A
1001-4020(2017)05-0590-04
10.11973/lhjy-hx201705023
2016-05-11
国家重大科学仪器设备开发专项(2012YQ090229);海南省自然科学基金(20152020)
赵振东(1981-),男,天津人,实验师,主要从事色谱、质谱分析工作,zhaozhendong@hainu.edu.cn
猜你喜欢
中学生数理化·高一版(2022年4期)2022-05-09
当代水产(2022年2期)2022-04-26
中国饲料(2021年17期)2021-11-02
山东第一医科大学(山东省医学科学院)学报(2021年7期)2021-10-13
当代水产(2021年2期)2021-03-29
昆明医科大学学报(2020年12期)2021-01-26
山东化工(2019年11期)2019-06-26
浙江大学学报(工学版)(2016年9期)2016-06-05
烟草科技(2015年8期)2015-12-20
中国药理学与毒理学杂志(2015年3期)2015-12-16
