
玉米制品中黄曲霉毒素的柱前和柱后衍生方法对比
2017-07-18 11:34王敏李广益邢燕李婷婷王勤
食品研究与开发 2017年14期
王敏,李广益,邢燕,李婷婷,王勤
(淄博市疾病预防控制中心,山东淄博255026)
玉米制品中黄曲霉毒素的柱前和柱后衍生方法对比
王敏,李广益,邢燕*,李婷婷,王勤
(淄博市疾病预防控制中心,山东淄博255026)
玉米制品中黄曲霉毒素测定的2种衍生化方法(柱前衍生和柱后衍生)进行比较。样品经过MycrosepTM226多功能净化柱净化,采用Waters Symmetry C18(4.6 mm×250 mm,5.0 μm)色谱柱进行分离,荧光检测器检测。柱前衍生法:样品提取液经净化后,采用三氟乙酸进行衍生后测定。柱后碘衍生法:样品提取液经净化后,经高效液相色谱分离、柱后碘衍生,荧光检测器检测。结果表明:柱前衍生法,黄曲霉毒素G1、B1检出限为0.05 μg/kg,黄曲霉毒素G2、B2检出限为 0.015μg/kg,r>0.999 1,回收率在 75.4%~84.2%之间;柱后碘衍生法,黄曲霉毒素 G1、B1检出限为 0.08 μg/kg,黄曲霉毒素G2、B2检出限为0.015 μg/kg,r>0.999 2,回收率在75.2%~85.7%之间。2种衍生方法在线性范围、精密度、回收率等方面较相似,表明柱前衍生法和柱后碘衍生法均适用于玉米制品中的黄曲霉毒素检测。
黄曲霉毒素;柱前衍生;柱后衍生;对比
黄曲霉毒素是黄曲霉菌和寄生曲霉在一定温度、湿度条件下产生的真菌毒素,世界卫生组织的国际癌症研究机构于1993年将黄曲霉毒素划定为Ⅰ类致癌物。常见的黄曲霉毒素有 B1、B2、G1、G2,其中 B1的毒性和量均为最大。我国GB 2761-2011《食品安全国家标准食品中真菌毒素限量》中规定玉米及其制品中的黄曲霉毒素B1限量标准为20 μg/kg,但标准尚未列出食品中黄曲霉毒素 B2、G1、G2的限量规定[1]。因此,有必要建立稳定可靠、灵敏度高的方法,为建立食品中黄曲霉毒素的限量标准提供依据。
我国食品中黄曲霉毒素 B1、B2、G1、G2的检测方法主要有薄层色谱法[2]和高效液相色谱法(HPLC)[3-4]以及液相色谱-质谱联用法[5-6]。其中,HPLC法因为其方法准确、灵敏,被广泛采用。HPLC法检测黄曲霉毒素,需衍生提高其荧光强度。目前,常用的衍生化方法有柱前衍生法、柱后卤衍生法、柱后紫外光衍生法以及柱后电化学衍生法等方法。本研究选取玉米制品作为研究样品,样品经过前处理后,分别用2种衍生化方法(柱前衍生法和柱后碘衍生法)进行衍生,荧光检测器检测,并对2种衍生化方法进行了验证和比较。
1 试验部分
1.1 仪器与试剂
e2695型高效液相色谱仪配荧光检测器、Waters Symmetry C18(4.6 mm×250 mm,5.0 μm)色谱柱:Waters公司;乙腈(色谱纯):迪马;多功能净化柱:MycrosepTM226柱:Romer Labs公司;混合标准贮备液为B1:1.0 μg/mL,B2:0.3 μg/mL,G1:1.0 μg/mL,G2:0.3 μg/mL,贮存温度:-10℃,定值单位:美国O2Si smart solutions,有效期至2017年3月。混合标准应用液浓度为B1:0.10μg/mL,B2:0.03μg/mL,G1:0.10μg/mL,G2:0.03μg/mL。
1.2 色谱分离条件及衍生条件
色谱柱:Waters Symmetry C18(4.6 mm×250 mm,5.0 μm);柱温:40 ℃;激发波长:360 nm,发射波长:440 nm;进样体积:20 μL。
1.2.1 柱前衍生法
梯度洗脱程序见表1。

表1 梯度洗脱程序Table 1 Gradient elution conditions
1.2.2 柱后碘衍生法
以甲醇-乙腈-水(25∶10∶65,体积比)为流动相,流速0.8 mL/min;衍生溶液为0.05%碘溶液(取碘0.5 g,加入甲醇100 mL使溶解,用水稀释定容至1 000 mL,即得),流速0.5 mL/min,衍生反应温度70℃。
1.3 标准曲线的制备
1.3.1 柱前衍生法
准确吸取混合标准应用液 1、2、10、20、100、200 μL于10 mL刻度吸管中,氮气挥干。加入200 μL正己烷和 100 μL 三氟乙酸,混匀 30 s后,(40±1)℃水浴衍生15 min,转移至1.5 mL样品瓶中,氮气吹干后用乙腈-水(15∶85,体积比)定容至 1.0 mL,进样 20 μL,测定峰面积,以峰面积Y为纵坐标,标准溶液浓度(ng/mL)X为横坐标,绘制标准曲线。
1.3.2 柱后碘衍生法
准确吸取混合标准应用液 1、2、10、20、100、200 μL于1.5 mL样品瓶中,用乙腈-水(15∶85,体积比)定容至1.0 mL,进样20 μL,测定峰面积,以峰面积Y为纵坐标,标准溶液浓度(ng/mL)X为横坐标,绘制标准曲线。
1.4 样品液的制备
1.4.1 柱前衍生法
准确称取试样5.0 g左右于50 mL离心管,加入20 mL乙腈-水(84∶16,体积比)提取,5 000 r/min离心5 min,移取约8 mL提取液经过功能净化管净化后,从收集池内准确移取2 mL净化液于棕色具塞小瓶中,60℃水浴下氮气吹干,加入200 μL正己烷和100 μL三氟乙酸,混匀 30 s后,(40±1)℃水浴衍生 15 min。氮气吹干,以1 mL乙腈-水(15∶85,体积比)溶解,混匀30 s,过 0.22 μm 水膜,待测。
1.4.2 柱后碘衍生法
准确称取试样5.0 g左右于50 mL离心管,加入20 mL乙腈-水(84∶16,体积比)提取,5 000 r/min离心5 min,移取约8 mL提取液经过功能净化管净化后,从收集池内准确移取2 mL净化液于棕色具塞小瓶中,60℃水浴下氮气吹干,以1 mL乙腈-水(15∶85,体积比)溶解,混匀 30 s,过 0.22 μm 水膜,待测。
2 结果与分析
2.1 色谱图
柱前衍生法和碘柱后衍生法的色谱图见图1。


图1 柱前衍生法(A)和柱后碘衍生法(B)HPLC色谱图Fig.1 HPLC chromatograms of pre-column derivatization(A)and post-column derivatization(B)
2.2 标准曲线及检出限
线性范围及检出限见表2。

表2 黄曲霉毒素的回归方程、相关系数及线性范围(柱前衍生法和柱后碘衍生法)Table 2 Regression equations,correlation coefficients and linear ranges of aflatoxins(pre-columnderivatization and post-column derivatization with iodine)
结果表明:柱前衍生法,黄曲霉毒素G1、B1检出限为 0.05 μg/kg,在 0.15 μg/kg~30 μg/kg 范围内线性关系良好,黄曲霉毒素 G2、B2检出限为 0.015 μg/kg,在0.05 μg/kg~10 μg/kg 范围内线性关系良好;柱后碘衍生法,黄曲霉毒素 G1、B1检出限为 0.08 μg/kg,在0.25 μg/kg~50 μg/kg 范围内线性关系良好,黄曲霉毒素 G2、B2检出限为 0.015 μg/kg,在 0.05 μg/kg~10 μg/kg范围内线性关系良好。
2.3 方法回收率和精密度
在已知不含黄曲霉毒素的玉米制品中分别加入10、100 μL混合标准应用液,按1.4节样品液的制备试验方法进行提取、净化和检测,重复测定6次,计算各种黄曲霉毒素的平均回收率及相对标准偏差,其结果见表3。
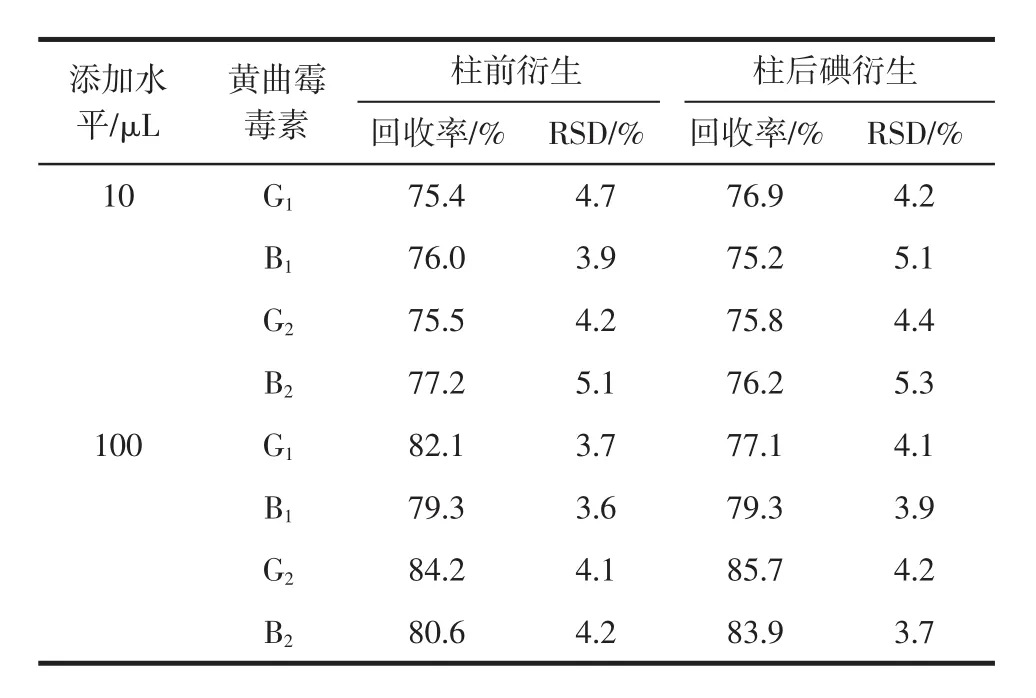
表3 方法回收率及精密度(n=6)Table 3 Recoveries and precision
2.4 样品测定
采用上述2种方法分别对市售10份玉米制品进行测定,均未检出黄曲霉毒素。
3 结论与讨论
本研究采用两种衍生化方法对玉米制品中的黄曲霉毒素进行了检测,并对两种方法进行了比较。结果表明柱前衍生和柱后碘衍生法测定黄曲霉毒素在线性范围、重复性、回收率等方面较相似,两种方法均适用于玉米制品中的黄曲霉毒素检测。柱前衍生法无需专门的柱后衍生装置,但前处理过程中需要对样品进行衍生,较为繁琐;碘衍生化法需要添加专门的柱后衍生泵来输送衍生液,而且每天都需要配制腐蚀性的碘溶液。柱前衍生法对于小批量的样品比较适用,柱后衍生法在大批量样品时候可以节省大量的人力,具体工作时,可根据样品量的大小以及实验室仪器的配备情况等选择合适的方法。
[1]中华人民共和国卫生部.GB 2761-2011食品安全国家标准食品中真菌毒素限量[S].北京:中国标准出版社,2011
[2]柳其芳.酶联免疫吸附法和薄层色谱法联合分析黄曲霉毒素B1的研究[J].中国热带医学,2006,6(2):246-248
[3]李军,于一茫,田苗,等.免疫亲和柱净化-柱后光化学衍生-高效液相色谱法同时检测粮谷中的黄曲霉毒素、玉米赤霉烯酮和赭曲霉毒素 A[J].色谱,2006,24(6):581-584
[4]冯靓,蔡增轩,谭莹,等.HPLC同时测定食品中黄曲霉毒素B1、B2、G1、G2[J].中国卫生检验杂志,2007,17(3):511-513
[5]宫小明,任一平,董静,等.超高效液相色谱串联质谱法测定花生、粮油中18种真菌毒素[J].分析测试学报,2011,30(1):6-12
[6]朱聪英,应永飞,韦敏钰.液相色谱-串联质谱法测定饲料中黄曲霉毒素的研究[J].质谱学报,2010,31(4):240-246
Comparison of Pre-column Derivatization and Post-column Derivatizationfor Aflatoxin in Corn Products
WANG Min,LI Guang-yi,XING Yan*,LI Ting-ting,WANG Qin
(Zibo Center for Disease Control and Prevention,Zibo 255026,Shandong,China)
To compare 2 derivatization methods for determination of aflatoxin in corn products(pre-column derivatization and post-column derivatization).The samples were purified by MycrosepTM226 multifunction purification column,separated by Waters Symmetry C18 (4.6 mm×250 mm,5.0 μm)and the fluorescence detector was used.Pre-column derivatizationmethod:sample extraction solution was determined by the use of three fluorine acetic acid for derivatization after purification.Post-column derivatization with iodine:the sample extract was purified by high performance liquid chromatography,and then detected by fluorescence detector.For pre-column derivatization,the detection limit of aflatoxin G1,B1was 0.05 μg/kg,the detection limit of aflatoxin G2,B2was 0.015 μg/kg ,r>0.999 1,and the recovery rate was between 75.4%and 84.2%;for post-column derivatization,the detection limit of aflatoxin G1,B1was 0.08 μg/kg,the detection limit of aflatoxin G2,B2was 0.015 μg/kg ,r>0.999 2,and the recovery rate was between 75.2%and 85.7%.The 2 methods are similar in terms of linear range,precision,recovery and so on.The results showed that the pre-column derivatization and post-column derivatization are all suitable for the detection of aflatoxin in corn products.
aflatoxin;pre column derivatization;post column derivatization;comparison
2016-10-17
10.3969/j.issn.1005-6521.2017.14.030
山东省自然科学基金项目(ZR2015PH035);山东省医药卫生科技发展计划项目(2013WS0028)
王敏(1985—),女(汉),主管技师,硕士研究生,研究方向:公共卫生理化检验。
*通信作者:邢燕,主管技师。
猜你喜欢
化工设计通讯(2022年10期)2022-12-31
波谱学杂志(2022年2期)2022-06-14
食品安全导刊(2021年21期)2021-08-30
中成药(2021年5期)2021-07-21
现代畜牧科技(2021年2期)2021-03-19
天然产物研究与开发(2018年9期)2018-10-08
中成药(2017年6期)2017-06-13
现代检验医学杂志(2015年1期)2015-02-06
中成药(2014年7期)2014-02-28
云南畜牧兽医(2014年4期)2014-02-28
