
Cellular immunity augmentation in mainstream oncologic therapy
2017-07-18 11:08DaohongChenXiaoshiZhangResearchInstituteofBiomedicineYilingPharmacyShijiazhuang050035ChinaStateKeyLaboratoryofOncologyBiotherapyCenterSunYatsenUniversityCancerCenterStateKeyLaboratoryofOncologyinSouthChinaCollaborativeInn
Cancer Biology & Medicine 2017年2期
Daohong Chen, Xiaoshi ZhangResearch Institute of Biomedicine, Yiling Pharmacy, Shijiazhuang 050035, China;State Key Laboratory of Oncology, Biotherapy Center, Sun Yat-sen University Cancer Center; State Key Laboratory of Oncology in South China; Collaborative Innovation Center for Cancer Medicine, Guangzhou 50060, China
Cellular immunity augmentation in mainstream oncologic therapy
Daohong Chen1, Xiaoshi Zhang21Research Institute of Biomedicine, Yiling Pharmacy, Shijiazhuang 050035, China;2State Key Laboratory of Oncology, Biotherapy Center, Sun Yat-sen University Cancer Center; State Key Laboratory of Oncology in South China; Collaborative Innovation Center for Cancer Medicine, Guangzhou 510060, China
Anticancer immunotherapy has undergone a long evolving journey for decades, and has been dramatically applied to mainstream treatments in oncology in recent 5 years. This progress represents an advanced milestone following cytotoxic medicine and targeted therapy. Cellular immunity plays a pivotal role in the immune responses of hosts to tumor antigens. Such immunity is notably suppressed during neoplastic progression due to immuno-editing processes. Cellular immunity can also be selectively reactivated to combat malignancies while exploiting the advantages of contemporary scientific breakthroughs in molecular immunology and genetic engineering. The rapid advancement of cellular immunity-based therapeutic approaches has achieved high efficacy in certain cancer patients. Consequently, the landscape of oncologic medicine and pharmaceutical innovation has transformed recently. In this regard, we present a comprehensive update on clinically established anti-cancer treatments with cell immunity augmentation as the major mechanism of action.
Cellular immunity; oncology; pharmaceutical innovation
Introduction
Biomedical background
The concept of immuno-surveillance was initially conceived four decades ago. Since that time, the roles of immune cells in tumor pathogenesis have been extensively investigated in various biomedical fields. In recent years, these roles were innovatively manipulated to improve clinical outcomes in cancer patients1,2. Considering the advantages of the breakthroughs in cell biology, protein chemistry, molecular oncology, and genetic engineering, immunological approaches have undergone a long evolving journey. These approaches have been increasingly applied in mainstream oncologic therapy. This progress thereby represents an advanced milestone following cytotoxic chemotherapy and targeted agents in the history of pharmaceutical treatments against cancer3,4. From a scientific viewpoint, the emergence of immuno-oncology re-defines cancer as a comprehensive disease involving multiple body systems. This definition transcends the traditional notion of cancer as a bulk of malignant cells in a local microenvironment1,5. Clinically, manipulating cellular immunity-based pharmaceutical intervention has enhanced the likelihood of successfully transforming malignant disorders into a group of manageable medical conditions, such as other chronic diseases, in several manners. This result is achieved because checkpoint inhibitors are reportedly capable of affording a long-term survival benefit of nearly 10 years to certain cancer patients6.
Immuno-oncology
The physiological immune network serves as the body’s defense system for eliminating etiological identities, including pathogenic microbes and tumor cells, by recognizing foreign antigens expressed in these cells1,2. To date, tumorspecific antigens remain to be well delineated. However, several tumor-associated self-antigens have been exploited to confer an acceptable safety window for immunotherapy against cancer3,7. In vivo anti-tumor responses are principally mediated by two arms of the cellular immunity, namely, innate and adaptive compartments in the immune system1,6. Innate immunity against neoplasms immediately occurs when tumor cells are detected, and recruits natural killer(NK) cells to play a pivotal role8. By contrast, the adaptive anti-tumor immune responses are processed in a more complex manner, mainly depending on antigen-presenting cells (APC) and T-lymphocytes, such as CD8+or CD4+T cells1,2. Furthermore, the interactions between neoplastic cells and the immune system have been dynamically dissected into a patho-biological progressing course of threephases on the base of immuno-editing theory6,9. In this sense, the elimination phase defines an ideal immuno surveillance action wherein initially transformed cells are cleared by the body’s defense system upon tumor antigen detection. Subsequently, in the equilibrium phase, the immune system gradually loses domination and allows neoplastic cells to survive in a dormant state2. This phenomenon is caused by a buildup of balance between opposing forces that develop from the tumor microenvironment10. Finally, in the escape phase, cancer cells outgrow beyond the controlling capacity of the host immune system resulting from the selective rise of less immunogenic and apoptosis-resistant malignant cells. In parallel, there is a locally elevated secretion of immunesuppressing factors, such as transforming growth factor-β (TGF-β) and vascular endothelial growth factors (VEGF), which are associated with the expansion of regulatory T (Treg) cells and myeloid-derived suppressor cells (MDSC)2,10. In addition, immuno-inhibitory checkpoint molecules, including cytotoxic T-lymphocyte-associated protein-4 (CTLA-4) and programmed cell death-1 (PD-1), have emerged as a group of important contributors to immune escape during cancer progression in recent years7,10.
The concept of biological therapy against neoplasm by intensifying immunesurveillance was proposed decades ago2. Even so, immune-cell-modulation-based strategies have remained outside mainstream therapy in clinical oncology until recent years. Through numerous long-term efforts, the renaissance of immunotherapy has finally reached contemporary oncology, and has been able to deliver impressive therapeutic benefits to certain cancer patients beyond chemotherapy and targeted regimens3,7. Herein, we highlight a systematic update on the successful clinical therapeutic approaches based on the augmentation of cellular immunity to control cancer (Figure 1, Table 1).
Adoptive cellular immunotherapy (ACIT)
Basic concept
ACIT is based on the notion that autologous immune cells acquire preferable anti-tumor potentials after exposure to neoplasms in the host bodies. This concept can be exploited for eradicating primary and metastatic cancer cells through biologically manipulated processes2. In principle, immune cells are isolated from the peripheral blood or tumor tissues of patients,ex vivoexpanded or with antigen stimulation/conditional medium selection, and then re-infused back into the patients3. Interestingly, immunotherapy with expanded activated autologous lymphocytes was revealed to remarkably up-regulate CD3+CD8+cells while diminishingCD4+CD25+cells in gastric cancer patients at late stages. As a result, overall survival (OS) is significantly extended for 4 months10,11.
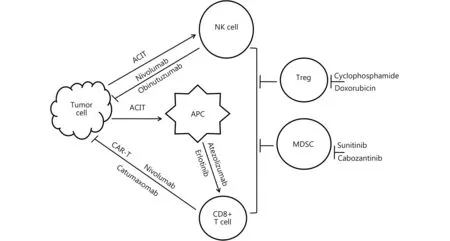
Figure 1Anti-cancer medicine with cellular immune mechanism. ACIT: adoptive cellular immunotherapy. APC: antigen presenting cell. CAR-T: chimeric antigen receptor-T cell. MDSC: myeloid-derived suppressor cell. NK: natural killer. Treg: regulatory T-cell.

Table 1Anti-cancer medicine depending on cellular immunity
ACIT-derived cancer vaccine
In addition, the sipuleucil-T vaccine therapy extends beyond the classic ACIT, which comprises autologous peripheral blood mononuclear cells primedex vivo. A recombinant protein of prostatic acid phosphatase serves as the tumorassociated antigen, and granulocyte-macrophage colonystimulating factor is also used to augment tumor antigen presentation by driving the cellular differentiation toward APC phenotypes. Impressively, this comprehensive therapeutic approach has been approved by the U.S. Food & Drug Administration (FDA) of the USA to treat castrationresistant prostate cancer because of the significant improvement of OS by 4.1 months and the reduction of the death risk by 22%2,12.
Tumor-infiltrating-lymphocyte-based ACIT
ACIT is alternatively designed to utilize tumor-infiltrating lymphocytes (TILs), which mainly contain mixtures of CD8+and CD4+T cells derived from resected metastatic tumor tissue, to reverse the immune-suppressive situation in the tumor microenvironment. The clinical outcomes of this approach can be further improved through the antigenspecific T-cell receptor (TCR) gene engineering1. The outcomes may also be enhanced by selectively depleting certain subpopulations of immune cells, such as Treg and MDSCs, with shifting cytokine profiles in tumor milieus1,3,13. Of note, several recent clinical trials were conducted in patients with metastatic melanoma refractory to standard therapies. In these investigations, the autologous TIL re-infusion-based ACIT strategy conferred objective response rates ranging from 49% to 72%. This strategy also dramatically resulted in a complete tumor regression observed in 22% of the patients, which remained efficacious for up to 82 months14,15. However, despite the above mentioned results, the therapeutic efficacy of TIL-based ACIT remains to be verified for other tumor types beyond melanoma3,16. Chimeric antigen receptor (CAR) T cell therapy represents a highly advanced category of ACIT that breaks the critical restrictions in the processing of classic immune responses1,2. This therapeutic approach is separately discussed later in this paper.
Immune checkpoint blockade
CTLA-4 pathway
Antibody therapy has risen as a hallmark of biologicalmedicine. This therapy has played an increasingly important role in the mainstream treatments against cancer2. A therapeutic protein against cancer is traditionally designed to neutralize a signaling cascade driving malignant cell growth or tumor angiogenesis. By contrast, antibody-based agents are currently further explored in comprehensive contexts of disease biology, particularly the interaction between tumor cells and the immune system1,17. The concept of immune surveillance holds that the host defense network can potentially recognize tumor antigens, such as somatic gene mutations, during onco-pathogenesis. As a result, immune responses to cancer are sequentially mounted9. Unfortunately, these responses are usually inefficient in eliminating established neoplasms because of the immune-editing process, which is driven by inhibitory signaling activities, such as up-regulated CTLA-4 and PD-1 checkpoint pathways3,7. CTLA-4 is exclusively expressed on T-lymphocytes. It substantially contributes to the control of initiated immune responses in line with physiological homeostasis. CTLA-4 is often induced by cancer cells to suppress the anti-tumor immunity18. In this regard, ipilimumab, an anti-CTLA-4 antibody approved by the FDA as first-line therapy for metastatic melanoma, delivers impressive clinical benefits, including 10 years’ addition to OS in certain terminal-stage patients3,19. Nonetheless, additional effort is still necessary to render anti-CTLA-4 antibodies efficacious for a larger patient population with melanoma. The possible therapeutic responses of CTLA-4-targeted agents to other tumor types remain to be tested18.
PD-1 axis
Similarly, PD-1 and its ligand (PD-L1) represent a novel immune checkpoint pathway that has attracted increasing interest; this pathway is highly induced during neoplastic development17. PD-1/PD-L1 axis participates in immune regulation in peripheral tissues because of the extensive distribution of PD-1 and its ligand. Of note, PD-1 is present on T, B, and NK cells. By contrast, the expression of PD-L1 extends beyond several immune cell types to exist on microvascular endothelial cells and tumor cells18,20. In this regard, nivolumab, an anti-PD-1 antibody, was demonstrated to clinically produce an effective response in melanoma. Particularly a few patients were found free from disease progression for years and causing fewer autoimmunityassociated adverse events than those with ipilimumab17,20. Nivolumab is also approved for therapeutic application in non-small cell lung cancer and renal carcinoma. This application implies the extensive anti-tumor roles of the antibody6,18. Recently, anti-PD-L1 antibody atezolizumab has been approved by the FDA to treat advanced bladder cancer. This progress clinically validates a new tumor type responsive to the cellular-immunity-augmentation-based therapy17,21. As a result, the approved immune checkpoint-blocking antibodies are expected to be tested on a broader spectrum of cancer types to expand the antibody therapy’s clinical indications18. Given the success of the above-mentioned checkpoint-neutralizing antibodies in the clinic, new agents targeting novel immune co-regulatory pathways are actively explored to potentially deliver further therapeutic benefits to cancer patients. These novel pathways include lymphocyteactivation gene-3 (LAG-3), T cell immunoglobulin and mucin domain-3 (TIM-3), and OX40, among others6,22.
New-generation antibodies
Bispecific antibody
Therapeutic antibodies are well known to possess the desirable properties, such as prolonged half-lives in plasma and high molecule-targeting specificity with minimized offtarget toxicity, in comparison with those of small chemical compounds7. Nevertheless, several limitations exist in antibody agents. Such single-targeted therapy may be unable to ideally tackle severe diseases (i.e., cancer) which usually involve multiple biological pathways in their pathogeneses1,10. Certain groups of patients showed an initial impressive response to a single-pathway-blocking-based treatment. However, the majority of these patients relapsed due to drug resistance caused by target gene mutation(s) or/and alternative signaling pathway activation7,9. One possible approach to circumvent this challenge is the bispecific antibody (BsAb) therapy, which has recently achieved some clinical success23. Blinatumomab, a T-lymphocyte-engaging BsAb simultaneously binding CD3-positive cytotoxic T cells and CD19-positive malignant cells, has been approved to treat relapsed/refractory B-acute lymphoblastic leukemia. This approval is based on a 43% responsive rate in phase 2 clinical trial after two treatment cycles24. Catumaxomab, which targets both EpCAM and CD3, is clinically available for EpCAM-positive tumors with malignant ascites. The antibody is particularly effective in halting the peritoneal spread of gastrointestinal or gynecologic neoplasms23. Currently, over 20 therapeutic BsAbs exist in clinical trials. These antibodies are intended to exert further therapeutic benefits to patients with various tumor types17,23.
Fc-modified antibody
First-generation monoclonal antibodies (mAbs) are produced from mouse B-cell hybridomas; mAb can be recognized by the human body defense system as foreign proteins and induce immune rejection. This occurrence results in immunity-associated side effect and short half-life time in the plasma17. The second-generation therapeutic mAbs acquire high target-antigen affinity and low immunogenicity through genetic engineering to generate the antibody variants with human or humanized amino-acid sequences. These improvements helped solve the above mentioned problems in the first-generation-mAbs2. Recently, over 20 third-generation mAbs have been developed through Fc region modification or/and glyco-manipulation to augment cellular immunity, such as antibody-dependent cellmediated cytotoxicity (ADCC). This new technology fully utilizes the biological potentials of an entire antibody molecule in vivo2,8. For example, the V158 site of IgGFcRIIIa, and defucosylated/low fucosylated mAbs have been identified to bind to the Fc receptor of immune cells, such as NK cells, with high affinity; this binding induces a strong ADCC25,26. Mogamulizumab, a defucosylated anti-CC chemokine receptor 4 (CCR4) mAb, has achieved clinical success by resulting in an impressive therapeutic response rate and prolonging the OS of acute T-cell leukemia/lymphoma patients17. An Fc-region-modified anti-CD20 mAb obinutuzumab has also been approved to treat drug-resistant chronic lymphocytic leukemia/lymphoma. This mAb was revealed to deliver a higher response rate and longer progression-free survival than those from the old-generation mAb rituximab27. Thus, the superiority of therapeutic mAbs to small chemical compounds not only leads to the inhibition of the signaling pathways driving tumor growth, but also simultaneously promotes several components of the body’s defense system to fight cancer2. Comprehensive characterization of the functional sites and post-translational situations of the IgG Fc region could inspire the development of additional third-generation mAbs that would deliver further clinical benefits beyond those of the secondgeneration mAbs17,28.
CAR-T cells
Scientific progress and clinical efficacy
Although autologous TIL re-infusion afforded substantial therapeutic benefit to melanoma patients, the approach using conventional TILs was generally ineffective for other tumor types in the clinic3. To overcome this limitation, autologous T lymphocytes may be driven to over-express tumor-antigenspecific TCR by genetic engineering. These TCRs are aimed to recognize tumor antigens in the complexes of human leukocyte antigens (HLAs) usually down-regulated in cancer cells in progressed phases2,29. In this regard, a novel CAR-T cell strategy was designed to recognize tumor antigens in a non-HLA-dependent manner. This type of recognition was achieved through genetically engineered autologous T cells that express cancer-antigen-specific immunoglobulin-based fusion protein3,30. The signaling of first-generation CARs was mediated by CD3 chains only. By contrast, the secondgeneration CAR-T cells are extensively applied clinically because of the addition of a signaling domain of a costimulatory molecule such as CD2830,31. Currently, CAR-T cells targeting over a dozen of tumor antigens have been investigated for therapeutic effectiveness against cancer in numerous clinical trials. In these studies, CD19 in B-cell malignancies appeared as the most attractive tumor antigen targeted by this approach3,30,32. CD19-re-directed CAR-T cells achieved a complete remission (CR) rate ranging from 70% to 90% in the clinical trials for patients with resistant or relapsed acute lymphoblastic leukemia30. CD19-CAR-T cells also achieved a response rate of 57% in patients with endstage advanced chronic lymphocytic leukemia, among which a few cases remained in CR without relapse for over 4 years32.
Limitations
CAR-T cell-based protocols have achieved dramatic successes in managing different hematological malignancies, including lymphocytic leukemia, lymphoma, and multiple myeloma. Unfortunately, most clinical trials that utilized this approach to treat solid tumors failed to achieve therapeutic efficacy mainly because of T-cell trafficking obstacles and the immunosuppressive microenvironment10,30,33. Hence, scientists proposed to circumvent the challenges arising from solid neoplasms by arming CAR-T cells with expressed promigration chemokine receptors and combining with checkpoint inhibitor blockade33,34. Safety concerns regarding CART therapy were noted, but these concerns were usually manageable or reversible. Ideally, on-target, off-tumor toxicity can be prevented by selecting highly specific tumor antigens that are not expressed in normal tissues at all3. Realistically, CD19, as a tumor-associated antigen, is expressed in neoplastic and normal B-cells. This expression pattern explains the CD19-CAR-T-cell-induced B-cell aplasia, which requires immunoglobulin replacement and long-term follow-up30. Moreover, although cytokine releasesyndrome can be mitigated with the interleukine-6-blocking antibody, neurologic toxicities, without known mechanisms, are still self-limited over several days in most cases30,35.
Classic therapy extended to boost anti-cancer immunity
Conventional medicine
Historically, a consensus exists on the design of chemotherapeutic compounds to kill fast-proliferating cancer cells by inhibiting DNA synthesis or perturbing mitosis. However, this approach may collaterally damage the normal cell of rapid dividing, and thus result in a few severe adverse events, such as immune activity suppression7. Nonetheless, combining immunotherapy with sequential or concurrent chemotherapy was serendipitously observed to eventually elicit a good clinical response in patients with advanced cancer under certain circumstances10,36. Thus, several possible mechanisms behind the synergistic phenomenon have been proposed, including improved antigen presentation, sensitized immunogenic cell death (ICD) induction, and minimized inflammatory activities in the tumor microenvironment. Furthermore, down-regulated suppressor cells, such as MDSC and Treg, have been recently noted to be clinically relevant and predictive of patient survival36. In particular, low dosages of cyclophosphamide and doxorubicin were revealed to successfully eliminate Treg and MDSC, respectively, in tumor-infiltrating lymphocytes. Hence, the anti-cancer efficacy of NK and CD8+T cells were enhanced10,37,38. Numerous clinical trials are currently underway to potentially enhance the ICD activity through exploiting conventional chemotherapeutic medicines. These pursuits often result in immunological responses linked to clinical benefits in cancer patients3,36,39. For example, the clinical efficacy of ACIT using in vitro expanded TIL in melanoma patients appears to be dependent on its prior condition under chemotherapeutic manipulation36.
Targeted agents
Over the last two decades, dramatic breakthroughs in cellular and molecular biology have clarified the delineations among novel signaling pathways that control proliferation, cell death/differentiation, angiogenesis, and metabolism7. As a result, this scientific progress caused innovative drug research and development, to fundamentally transform toward targeted therapy, particularly in oncology7,40. Selective blocking is crucial to signaling pathways driving tumor growth. By doing so, targeted therapeutic agents show high clinical efficacy and minimized adverse effects compared with those of conventional medicine. Interestingly, several targeted medicines have been found to positively influence diverse aspects of cellular immunity against cancer in recent years17,40. Besides suppressing malignant cell proliferation and angiogenesis in the tumor microenvironment, the kinase inhibitors sunitinib and cabozantinib can improve therapeutic efficacy by blocking the signal transducer and activator of transcription 3 (STAT3) to diminish Treg/MDSCs and increase the number of CD8+cells10,40,41. In parallel, epidermal-growth-factor-receptor-targeted medicines erlotinib and cetuximab enhanced major histocompatibility complex expression, and thus augmented tumor antigen presentation and ICD40,42. In this regard, numerous on-going clinical trials combine immune treatments with these targeted agents to boost anti-tumor immunity, and achieve therapeutic efficacy in cancer patients40,43.
Perspective
Although the potential of anti-tumor immunity was recognized over a century ago, immunotherapy in oncology has not been able to significantly improve the clinical endpoints of cancer patients. Such improvement has been achieved only in recent years when contemporary pharmaceutical innovation is being deeply inspired by dramatic breakthroughs in biomedical sciences, particularly in cellular/molecular immunology and genetic engineering1,2. Checkpoint inhibitors and CAR-T approaches represent the hallmark accomplishments and promoted the inclusion of cellular immunity augmentation into mainstream oncologic therapy. Meanwhile scholars also noted a few practical limitations in the field, such as low responsive rate or few sensitive tumor types which hinder the extension of efficacy to an increased number of cancer patients3,22,32. Accordingly, efficacious rates for a given tumor type were improved via new synergistic strategies. In this sense, combining PD-1 antibodies and other immune co-regulators or cancer vaccines appeared to deliver better results. Moreover, various immunotherapeutics, plus certain conventional medicines, are in ongoing clinical trials17,36,39,40. In addition, to enhance the effectiveness of CAR-T therapy in providing clinical benefits to patient with solid tumors other than hematological malignancies, scientists proposed the augmentation of T-cell migration and manipulation of tumor microenvironment as promising strategies2,33,34.
While immunotherapy exhibits anti-tumor responses, itfrequently induces auto-immunity-based adverse effects that may collaterally damage body organs. Most known tumor antigens are tumor-associated antigens expressed in neoplastic and normal cells1,2. To address this challenge, further efforts are still necessary in terms of identifying novel tumor-specific antigens, such as growth signaling gene mutations that drive the therapeutic resistance to targeted medicines1,3. Cancer presents a highly dynamic and complex disease with inter-and intra-tumor heterogeneity; a few malignant cell subsets may exhibit low immunogenicity and are thus insensitive to systemic immunotherapy2,44. In this regard, a recent novel in situ immunotherapy can deliver advanced clinical benefits to cancer patients at late stages. This effect is achieved by the killing of malignant cells to release entire tumor antigens in a major cancer tissue using certain physical or chemical means. This step is followed by the local injection of an immune adjuvant to boost antigen presentation45,46. Hence, the insights herein remind that despite the advancement of systemic immunotherapy in oncology, local precision treatments should not be ignored to clinically fulfill the beneficial potentials in solid tumors.
Conflict of interest statement
No potential conflicts of interest are disclosed.
1.Miller JF, Sadelain M. The journey from discoveries in fundamental immunology to cancer immunotherapy. Cancer Cell. 2015; 27: 439-49.
2.Goel G, Sun WJ. Cancer immunotherapy in clinical practice -- the past, present, and future. Chin J Cancer. 2014; 33: 445-57.
3.Farkona S, Diamandis EP, Blasutig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016; 14: 73
4.Ascierto PA, Addeo R, Cartenì G, Daniele B, De Laurentis M, Ianniello GP, et al. The role of immunotherapy in solid tumors: report from the Campania Society of Oncology Immunotherapy (SCITO) meeting, Naples 2014. J Transl Med. 2014; 12: 291
5.Chen DH, Goswami CP, Burnett RM, Anjanappa M, Bhat-Nakshatri P, Muller W, et al. Cancer affects microRNA expression, release, and function in cardiac and skeletal muscle. Cancer Res. 2014; 74: 4270-81.
6.Pennock GK, Chow LQM. The evolving role of immune checkpoint inhibitors in cancer treatment. Oncologist. 2015; 20: 812-22.
7.Chen DH, Zhang XS. Targeted therapy: resistance and resensitization. Chin J Cancer. 2015; 34: 43
8.Rezvani K, Rouce RH. The application of natural killer cell immunotherapy for the treatment of cancer. Front Immunol. 2015; 6: 578
9.Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases--elimination, equilibrium and escape. Curr Opin Immunol. 2014; 27: 16-25.
10.Chen DH, Zhang XS. Tipping tumor microenvironment against drug resistance. Oncol Trans Res. 2015; 1: 106
11.Zhang GQ, Zhao H, Wu JY, Li JY, Yan X, Wang G, et al. Prolonged overall survival in gastric cancer patients after adoptive immunotherapy. World J Gastroenterol. 2015; 21: 2777-85.
12.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010; 363: 411-22.
13.Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005; 202: 907-12.
14.Yee C. Adoptive T-cell therapy for cancer: boutique therapy or treatment modality? Clin Cancer Res. 2013; 19: 4550-2.
15.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011; 17: 4550-7.
16.Li J, Chen QY, He J, Li ZL, Tang XF, Chen SP, et al. Phase I trial of adoptively transferred tumor-infiltrating lymphocyte immunotherapy following concurrent chemoradiotherapy in patients with locoregionally advanced nasopharyngeal carcinoma. Oncoimmunology. 2015; 4: e976507
17.Chen D. Emerging highlights of antibody therapy in oncology. Clin Oncol. 2016; 1: 1026
18.Quezada SA, Peggs KS. Exploiting CTLA-4, PD-1 and PD-L1 to reactivate the host immune response against cancer. Br J Cancer. 2013; 108: 1560-5.
19.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010; 363: 711-23.
20.Sunshine J, Taube JM. PD-1/PD-L1 inhibitors. Curr Opin Pharmacol. 2015; 23: 32-8.
21.Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014; 515: 563-7.
22.Swart M, Verbrugge I, Beltman JB. Combination approaches with immune-checkpoint blockade in cancer therapy. Front Oncol. 2016; 6: 233
23.Fan GW, Wang ZJ, Hao MJ, Li JM. Bispecific antibodies and their applications. J Hematol Oncol. 2015; 8: 130
24.Benjamin JE, Stein AS. The role of blinatumomab in patients with relapsed/refractory acute lymphoblastic leukemia. Ther Adv Hematol. 2016; 7: 142-56.
25.Monnet C, Jorieux S, Souyris N, Zaki O, Jacquet A, Fournier N, et al. Combined glyco- and protein-Fc engineering simultaneously enhance cytotoxicity and half-life of a therapeutic antibody. mAbs. 2014; 6: 422-36.
26.Wang W, Erbe AK, Hank JA, Morris ZS, Sondel PM. NK cellmediated antibody-dependent cellular cytotoxicity in cancerimmunotherapy. Front Immunol. 2015; 6: 368
27.Gabellier L, Cartron G. Obinutuzumab for relapsed or refractory indolent non-Hodgkin's lymphomas. Ther Adv Hematol. 2016; 7: 85-93.
28.Natsume A, Niwa R, Satoh M. Improving effector functions of antibodies for cancer treatment: Enhancing ADCC and CDC. Drug Des Dev Ther. 2009; 3: 7-16.
29.Chen DH, Radford-Smith G, Dipaolo MC, McGowan I, Jewell DP. Cytokine gene transcription of human colonic intraepithelial lymphocytes costimulated with epithelial cells bearing HLA-DR and its inhibition by 5-aminosalicylic acid. J Clin Immunol. 1996; 16: 237-41.
30.Almåsbak H, Aarvak T, Vemuri MC. CAR T cell therapy: A game changer in cancer treatment. J Immunol Res. 2016; 2016: 5474602
31.Chen DH, Iijima H, Nagaishi T, Nakajima A, Russell S, Raychowdhury R, et al. Carcinoembryonic antigen-related cellular adhesion molecule 1 isoforms alternatively inhibit and costimulate human T cell function. J Immunol. 2004; 172: 3535-43.
32.Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015; 7: 303r
33.Zhang H, Ye ZL, Yuan ZG, Luo ZQ, Jun HJ, Qian QJ. New strategies for the treatment of solid tumors with CAR-T cells. Int J Biol Sci. 2016; 12: 718-29.
34.Liu XJ, Ranganathan R, Jiang SG, Fang CY, Sun J, Kim J, et al. A chimeric switch-receptor targeting pd1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Res. 2016; 76: 1578-90.
35.Dai HR, Wang Y, Lu XC, Han WD. Chimeric antigen receptors modified T-cells for cancer therapy. J Natl Cancer Inst. 2016; 108: djv439
36.Andersen MH, Junker N, Ellebaek E, Svane IM, Thor Straten P. Therapeutic cancer vaccines in combination with conventional therapy. J Biomed Biotechnol. 2010; 2010: 237623
37.Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, et al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother. 2007; 56: 641-8.
38.Alizadeh D, Trad M, Hanke NT, Larmonier CB, Janikashvili N, Bonnotte B, et al. Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T-cell transfer in breast cancer. Cancer Res. 2014; 74: 104-18.
39.Vacchelli E, Aranda F, Eggermont A, Galon J, Sautès-Fridman C, Cremer I, et al. Trial Watch: Chemotherapy with immunogenic cell death inducers. Oncoimmunology. 2014; 3: e27878
40.Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. 2012; 12: 237-51.
41.Kwilas AR, Donahue RN, Tsang KY, Hodge JW. Immune consequences of tyrosine kinase inhibitors that synergize with cancer immunotherapy. Cancer Cell Microenviron. 2015; 2: e677
42.Im JS, Herrmann AC, Bernatchez C, Haymaker C, Molldrem JJ, Hong WK, et al. Immune-modulation by epidermal growth factor receptor inhibitors: implication on anti-tumor immunity in lung cancer. PLoS One. 2016; 11: e0160004
43.Antonia SJ, Larkin J, Ascierto PA. Immuno-oncology combinations: a review of clinical experience and future prospects. Clin Cancer Res. 2014; 20: 6258-68.
44.Chen D, Zhang X. Tackling cancer stem cell: from science to medicine. J Translational Clin Exp Oncol. 2016; 1: 6-11.
45.Zhou FF, Li XS, Naylor MF, Hode T, Nordquist RE, Alleruzzo L, et al. InCVAX--a novel strategy for treatment of late-stage, metastatic cancers through photoimmunotherapy induced tumor-specific immunity. Cancer Lett. 2015; 359: 169-77.
46.Gao F, Jing P, Liu J, Lu YF, Zhang PC, Han W, et al. Happenenhanced overall survival time in advanced hepatocellular carcinoma by ultro-minimum incision personalized intratumoral chemoimmunotherapy. J Hepatocellular Carcinoma. 2015; 2: 57-68.
Cite this article as: Chen D, Zhang X. Cellular immunity augmentation in mainstream oncologic therapy. Cancer Biol Med. 2017; 14: 121-8. doi: 10.20892/j.issn.2095-3941.2017.0022
Daohong Chen
E-mail: daohong@hotmail.com
February 28, 2017; accepted March 23, 2017. Available at www.cancerbiomed.org
Copyright © 2017 by Cancer Biology & Medicine
 Cancer Biology & Medicine2017年2期
Cancer Biology & Medicine2017年2期
- Cancer Biology & Medicine的其它文章
- Developmental pathways associated with cancer metastasis: Notch, Wnt, and Hedgehog
- Mesenchymal stromal cells’ role in tumor microenvironment: involvement of signaling pathways
- Simplified microsatellite instability detection protocol provides equivalent sensitivity to robust detection strategies in Lynch syndrome patients
- Significance of stromal-1 and stromal-2 signatures and biologic prognostic model in diffuse large B-cell lymphoma
- Immunohistochemical evaluation of vitamin D receptor(VDR) expression in cutaneous melanoma tissues and four VDR gene polymorphisms
- Concomitant-chemoradiotherapy-associated oral lesions in patients with oral squamous-cell carcinoma