
Developmental pathways associated with cancer metastasis: Notch, Wnt, and Hedgehog
2017-07-18 11:08ArmelHerveNwaboKamdjePaulTakamKamgaRichardTagneSimoLorellaVecchioPaulFaustinSekeEtetJeanMarcMullerGiulioBassiEriqueLukongRaghuveeraKumarGoelJeremieMboAmveneMauroKrampera
Cancer Biology & Medicine 2017年2期
Armel Herve Nwabo Kamdje, Paul Takam Kamga, Richard Tagne Simo, Lorella Vecchio, Paul Faustin Seke Etet, Jean Marc Muller, Giulio Bassi, Erique Lukong, Raghuveera Kumar Goel, Jeremie Mbo Amvene, Mauro Krampera
1Department of Biomedical Sciences, University of Ngaoundere, Ngaoundere 454, Cameroon;2Laboratory, University of Verona, Verona 37129, Italy;3Department of Basic, Health Sciences, College of Applied Medical Sciences, Qassim University, Buraydah 52571, Saudi Arabia;4Faculty of Science, University of Poitiers, Poitiers 86022, France;5Department of Biochemistry, College of Medicine, University of Saskatchewan, Saskatoon S7N 5E5, Canada
Developmental pathways associated with cancer metastasis: Notch, Wnt, and Hedgehog
Armel Herve Nwabo Kamdje1, Paul Takam Kamga2, Richard Tagne Simo1, Lorella Vecchio3, Paul Faustin Seke Etet3, Jean Marc Muller4, Giulio Bassi2, Erique Lukong5, Raghuveera Kumar Goel5, Jeremie Mbo Amvene1, Mauro Krampera2
1Department of Biomedical Sciences, University of Ngaoundere, Ngaoundere 454, Cameroon;2Laboratory, University of Verona, Verona 37129, Italy;3Department of Basic, Health Sciences, College of Applied Medical Sciences, Qassim University, Buraydah 52571, Saudi Arabia;4Faculty of Science, University of Poitiers, Poitiers 86022, France;5Department of Biochemistry, College of Medicine, University of Saskatchewan, Saskatoon S7N 5E5, Canada
Master developmental pathways, such as Notch, Wnt, and Hedgehog, are signaling systems that control proliferation, cell death, motility, migration, and stemness. These systems are not only commonly activated in many solid tumors, where they drive or contribute to cancer initiation, but also in primary and metastatic tumor development. The reactivation of developmental pathways in cancer stroma favors the development of cancer stem cells and allows their maintenance, indicating these signaling pathways as particularly attractive targets for efficient anticancer therapies, especially in advanced primary tumors and metastatic cancers. Metastasis is the worst feature of cancer development. This feature results from a cascade of events emerging from the hijacking of epithelial-mesenchymal transition, angiogenesis, migration, and invasion by transforming cells and is associated with poor survival, drug resistance, and tumor relapse. In the present review, we summarize and discuss experimental data suggesting pivotal roles for developmental pathways in cancer development and metastasis, considering the therapeutic potential. Emerging targeted antimetastatic therapies based on Notch, Wnt, and Hedgehog pathways are also discussed.
Cancer metastasis; developmental pathways; Notch; Wnt; Hedgehog; therapeutic targets
Introduction
The development of metastasis from a primary tumor site, including epithelial-mesenchymal transition (EMT), tumor neoangiogenesis, and spread of malignancy, is a multistep phenomenon for targeting tissues and organs. The spread of malignancy results from malignant cell transport through blood vessels to target tissues and organs, the invasion of the latter by infiltrating malignant cells, and the development of secondary tumors1,2. Master developmental pathways, such as Notch, Wnt, and Hedgehog, are signaling pathways that play pivotal roles along embryonic development. The role of Notch in solid tumor and hematological malignancy initiation and development has been extensively documented. In most cases, Notch is a major oncogene associated with tumor progression to metastasis, anoikis resistance, EMT, neoangiogenesis, malignant cell proliferation, and changes in tissue microenvironment promoting the homing of metastasis-promoting cells; nonetheless, Notch may also act as a tumor suppressor3-6.
The overexpression of Wnt signaling is common in many hematological malignancies and solid tumors. Clinical and experimental evidence suggests that Wnt/β-catenin activation is critical for cancer development, angiogenesis, migration, and invasion5-7. The antagonists of Wnt pathway, such as Wnt inhibitory factor 1 (WIF-1), Dickkopf proteins (Dkks), the secreted frizzled-related proteins (sFRPs), and Disheveled-axin domain containing 1 (DIXDC1), enhance the tumorigenic and metastatic processes of various cancer types in vitro and in vivo8-10. Similar to Notch and Wnt pathways, Hedgehog is an evolutionary and developmental pathway involved not only in cellular differentiation but also in physiological and tumorigenic control of postnatal cellular events, such as proliferation, cell death, motility, migration, and invasion5,6.
Emerging therapies targeting master developmentalpathways represent promising approaches for metastasis prevention and cancer stem cell elimination (Figure 1). In this study, we summarize the data supporting a role for developmental pathways in pro-metastatic processes, emphasizing on Notch, Wnt, and Sonic Hedgehog (Shh), considering their therapeutic potential. Emerging antimetastatic strategies targeting these developmental pathways are also discussed.
Pro-metastatic changes and developmental pathways
Resistance to anoikis
Similar to other anchorage-dependent cells, the survival of tumor cells, following the detachment from surrounding epithelial tissue or extracellular matrix, requires the capability to resist against the form of programmed cell death resulting from such detachment, namely, anoikis. Unlike most normal anchorage-dependent cells that undergo cell death when detached from the extracellular matrix, tumor cells can implement several molecular mechanisms, promoting their survival in suspension and the spread of metastases. The experimental and clinical evidence strongly suggests that the subversion of the repertoire of integrin developmental pathways is the main molecular mechanism favoring anoikis resistance in tumor cells.
Cell-cell and cell-extracellular matrix contacts support anchorage-dependent cell survival in an integrin-dependent manner, partly because of integrin-mediated activation of anti-apoptotic signaling pathways, such as phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt), mitogen-activated protein kinase (MAPK), and focal adhesion kinase pathways11,12. The aberrant constitutive activation of these anti-apoptotic signaling pathways results in post-translational modifications and changes in the levels of Bcl-2 antiapoptotic protein family members, i.e., multi-domain and BH3 domain-only proteins. Consequently, the overexpression of the latter proteins in tumor cells prevents the mitochondrial function disruptions that normally lead to programmed cell death13and related pro-apoptotic mechanisms, such as metabolism-induced autophagy11,12.
In addition to these intrinsic apoptotic mechanisms, metastatic competent cells also achieve resistance to anoikis and anticancer therapy by interfering with extrinsic apoptosis pathways, such as the families of death receptors, including Fas receptors13. Notably, the prevention of Fas receptor activity results in increased levels of cellular FADD-like IL-1β-converting enzyme-inhibitory protein, a master antiapoptotic regulator, in these tumor cells. This increase results in a cross talk between extrinsic and intrinsic caspase pathways that impair the capability to use common apoptotic caspases, particularly effector caspases, favoring tumor cell survival13. Integrin signaling has the capability to induce ligand-independent activation of developmental signaling molecules that are able to trigger resistance to anoikis. Examples include the loss in cell adhesion factors, such as E-cadherin; and the overactivation of other EMT signaling molecules, such as the mammalian target of rapamycin (mTORC1 and mTORC2)14-16, growth factors, such as epidermal growth factor (EGF) and vascular endothelial growth factor (VEGF) receptor signaling pathways12-15, and oncogenes, such as Wnt, β-catenin, Notch, and Shh17-20.
The potential of the pharmacological targeting of developmental signaling pathways favoring resistance to anoikis supports the survival of metastatic cells throughoutthe metastatic journey, i.e., from the primary site to the target tissue invasion and secondary tumor development12-16.
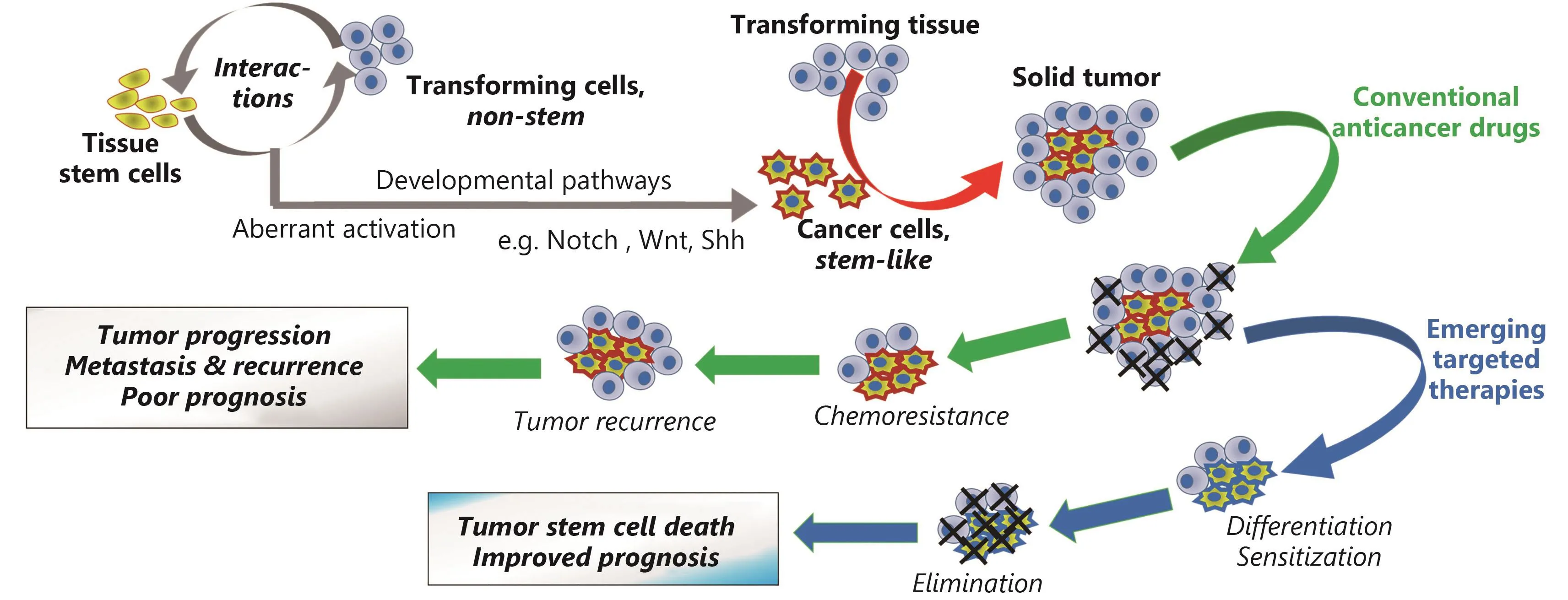
Figure 1Effects of conventional anticancer drugs and of their combination with emerging therapies targeting developmental pathways on cancer stem-like cells and treatment outcome.
EMT
EMT is a reversible specialized process that occurs during the normal embryonic development, which is pivotal for cell specialization and tissue patterning. During development, an early detachment of embryonic epithelial cell from the initial tissue occurs without activation of anoikis. Then, these cells acquire the motile shape that will allow them to migrate to new sites, where they will undergo mesenchymal-to-epithelial transition, providing these cells with the capability to build new tissues21,22. This process, which also occurs during wound healing, is used by transforming cells during tumorigenesis and tumor progression. Primary tumor cells undergo EMT during cancer progression, which confers them with a mesenchymal-like phenotype associated with not only high motility but also resistance to anoikis and anticancer drugs22-24.
While normal development of EMT is controlled and highly organized, this event is more stochastic in cancer25. Physiological EMT results from the coordination of molecular events with new cell capacity. Specifically, EMT starts by losing the cell junction proteins, such as E-cadherin, claudins, occludins, and catenins, associated with the epithelial organization, followed closely by the expression of mesenchymal markers, such as N-cadherin and vimentin26,27. A large number of oncogenic factors known to induce and/or to sustain tumor cell motility and metastasis, such as transforming growth factor beta (TGF-β) and insulin-like growth factor 1, induce the loss of the epithelial marker E-cadherin28,29. E-cadherin is at the center of EMT and anoikis signaling networks because its loss is common in cancer progression, where this marker is associated with aberrant EMT, tumor cell migration and invasion, and resistance to anoikis. Moreover, the loss of E-cadherin and other adheren junction molecules disrupts epithelial tissue, leading cells to the adoption of a minimally polarized phenotype with high invasive properties. The switch in cell morphologies and properties is mediated by key transcription factors, namely, EMT-activating transcription factors. Examples include the transcription factors of a snail family (e.g., snail, slug, and smuc), zinc-finger E-box-binding (ZEB) factors [e.g., the two-handed zinc-finger factors of d-crystallin/E2 box factor (dEF1) family proteins dEF1/ZEB homeobox 1 and Smadinteracting protein 1/ZEB2], and basic helix-loop-helix factors (e.g., Twist1, Twist2, E12, E47, and Tbx3)30-32.
Motility and migration
The metastatic competent cells detached from primary tumors require being migrated to invade other sites. The migration and other motility events result from specific cytoskeletal changes mediated and regulated mainly by small GTPase proteins of the Rho family33-36. The members of the Rho GTPase family process extracellular signal into information that will lead to the reorganization of the cytoskeleton. As discussed above, the molecular events that trigger EMT and confer anoikis can activate the migratory machine in tumor cells through the members of the Rho GTPase family35.
Mounting evidence supports Rho GTPase involvement in all tumorigenic events from the primary tumor initiation to the metastasis. These small GTPases direct actin dynamic and protrusive patterns in migrating cells, including the projection of the cytoskeletal protein actin on the leading edge of the cell (lamellipodia), the extension of slender cytoplasmic projections beyond the leading edge of lamellipodia (filopodia), and the extracellular matrix degradation-associated protrusions of the plasma membrane (invadopodia)36. Actin dynamic results from its polymerization induced by actin-related protein 2 (Arp2) and Arp3. Invadopodia is a characteristic of invasive tumor cells, whereas the other patterns can occur in normal processes37-40. Rho-A, Cdc42, and Rac1 are the most documented Rho GTPases involved in cell migration and metastasis. Besides these common properties, Rho-A is also linked to stress fiber, whereas Cdc42 and rac1 are involved in filopodia and lamellipodia, respectively37,38.
In addition to the EMT-induced protrusive processes, migrating tumor cells can undergo collective amoeboid transition and mesenchymal to amoeboid transition with characteristic invadopodia39,40. Notably, such invadopodia require the enzymatic digestion of the extracellular matrix by the leading edge, mediated by matrix metalloproteinases. This type of migration is observed in many solid tumors, including breast carcinoma41.
Neoangiogenesis
The angiogenic processes triggered during tumorigenesis, namely, neoangiogenesis, provide the neoplastic tissue with new blood vessels that will supply the high requirements of this tissue in nutrients and oxygen. Such high requirements are triggered by paracrine growth signaling aberrantly activated directly or indirectly in transforming cells in response to extracellular matrix digestion41,42. The new blood vessels are also used as highways by invading tumor cells,which are able to travel great distances in the body to start metastases in other organs because of the motility provided by EMT.
Neoangiogenic dynamic relies on angiogenic signaling molecules of the VEGF family (VEGF-A/B/C/D) and their receptors (VEGFR-1/2/3/4)43,44. The main activator of the angiogenic cascade in the transforming and neoplastic tissue is the hypoxic microenvironment. The low oxygen tension observed in the microenvironment induces the stabilization of hypoxia-inducible factors (HIFs), preventing the von Hippel-Lindau binding protein-mediated degradation of these transcription factors. Then, the HIFs enter the tumor cell nuclei, where they dimerize and associate with p/300 to form a transactivation complex. Consequently, this transcriptional complex will stimulate the expression of hypoxia target genes by binding to their hypoxia response elements. Hypoxia target genes include genes involved in the angiogenesis cascade, such as VEGF system genes, and other ligand-receptor systems with high angiogenic potential, such as angiopoetin/Tie2, platelet-derived growth factor (PDGF)/PDGF receptor, Delta-like ligand 4 (DLL4)/Notch, and fibroblast growth factor (FGF)/FGF receptor45-47.
Notch signaling and metastasis
Notch family of signaling molecules
The Notch family of the transmembrane proteins encompasses four receptors (Notch1-4) and five ligands (Jagged1/2, Dll-1/3/4). The Notch receptors are activated when they bind to a ligand expressed on the membrane of an adjacent cell. Then, the receptors undergo two proteolytic cleavages at S2 and S3 sites performed by ADAM10/17 metalloprotease and gamma secretase complex, respectively. The two proteolytic events result in the release of an intracellular active form of the receptors, namely, Notch intracellular domain (NICD). Subsequently, the NICD domains enter the nucleus, where they form a transcriptional activation complex with MALM1 and RBP-jk proteins. This complex will promote the expression of various genes involved in cell fate determination, including myc, cyclinD, and genes of the helix basic family, such as hes1 and hey1. Functionally, the products of this transcription will control the cell proliferation, cell death, adhesion, invasion, and migration.
Notch in the metastatic cascade
The survival pathways of Notch signaling controls are important for anoikis resistance, such as NF-κB, Akt, Sat3, and mTOR31. The capability to modulate the expression of the EMT transcription factors, such as snail, slug, ZEB1/2, TGF-β, FGF, and PDGF, increases the expression of the mesenchymal markers (e.g., slug), downregulates the epithelial markers, and activates the Rho GTPases3,18,48. For example, Kwon et al.7assessed the Notch signaling's prometastatic role in a mouse model of prostate cancer. The constitutive expression of the Notch1 intracellular domain in mouse prostate luminal cells (i) impaired secretory functions; (ii) suppressed anoikis via hes1 non-dependent NF-κB activity; and (iii) stimulated luminal cell proliferation by potentiating PI3K/Akt signaling. Recently, Shao and collaborators38emphasized the Jagged1-induced Notch signaling triggered migration, invasion, and a slug-dependent EMT transition in breast cancer cells. All these effects were abrogated by Notch silencing48, underlining the potential of Notch targeting for the modulation of tumor cell motility. Furthermore, Rho GTPase upregulation is common in patients diagnosed with T-cell acute lymphoblastic leukemia (T-ALL), a hematological malignancy, where Notch activating mutations are observed in more than 50% of the cases49.
Notch targeting as antimetastatic strategy
Notch-based therapeutic strategies
Notch-based therapeutic strategies were developed for various steps of the signaling cascade, including ligand-receptor interactions, enzymatic cleavages of the receptors, cytoplasmic interactions, and transcriptional activation complex. The requirement of S2 and S3 cleavages for the activation of Notch receptors made these events particularly attractive for the modulation of Notch-induced repression of E-cadherin, which associates with a number of pro-tumorigenic events and poor prognosis in solid cancers, such as breast cancer50. S2 and S3 are due to ADAM10/17 metalinebreak and secretase complex, respectively. Unlike S2 cleavage inhibitors, gamma secretase inhibitors (GSI) have been widely used in anticancer preclinical research, partly due to GSIs were developed first and tested in Alzheimer’s disease and experimental models, where they proved relatively safe51. GSIs achieving a total blockade of Notch signaling display strong antineoplastic responses, while even those achieving partial blockade can exhibit decent antineoplastic activities52,53.
GSI treatment mitigated the development of tumor cell invasion and metastasis in vivo and in vitro by preventing EMT, migration, invasion, and neoangiogenesis54. A numberof GSIs recently entered clinical trials, including BMS906024, MK0752, PF03084014, and R0492909755. PF03084014 is currently in phase 1 trials in metastatic pancreatic adenocarcinoma patients not previously treated with anticancer therapies. R04929097 is in phase 2 trials in metastatic melanoma56. MK0752 is in phases 1 and 2 trials for metastatic and advanced primary tumors, respectively, in breast cancer55. Moreover, blocking antibodies with GSI activity have also been developed. These agents target the components of GSI complex, such as nicastrin, the largest member of the complex. Antinicastrin monoclonal antibodies clone 2H6 elicited pleiotropic antimetastatic activities on invasive cancer cell lines, including an attenuation of invadopodia degradation of the extracellular matrix and delayed cancer cell extravasation through endothelial cells in the in vitro Boyden chamber invasion assay57.
The enthusiasm for anticancer and antimetastatic application of GSIs is mitigated by the limitation to gamma secretase targeting, considering that this Notch signaling component is not always critical in the pathological phenotype of malignancies. In addition, a number of tissues physiologically require Notch activity for tissue plasticity. Gastric epithelium is the most relevant example of such tissue. Its functional alterations partly explain the gastrointestinal toxicity observed following GSI treatment53,55,56. In vivo experiments suggest that currently, antinicastrin monoclonal antibodies are more potent than GSIs in clinical trials, with minimally marked gastrointestinal signs57. Collectively, these observations suggested the possibility to improve the therapeutic outcome of the targeting of Notch signaling by changing the specific targets and the approaches used. These findings raised the question of whether developing the inhibitors for specific Notch receptors or ligands would also improve therapeutic outcome.
Targeting Notch ligands and receptors
Notch ligand and receptor targeting is a particularly interesting approach because it allows specific targeting of Notch receptors or ligands critical in the pathological phenotype of malignancies, including the metastatic phenotype. For example, Notch1 was reported to control metastatic processes in small cell lung cancer cell lines and to initiate EMT and invasion of breast cancer cells48,58. The targeting of blocking monoclonal antibodies, specifically Notch receptor subtypes, has been developed. These molecules mainly act on the EGF-like repeats of Notch receptors. Various Notch receptor blocking antibodies have entered clinical trials for metastatic and advanced solid cancers. For example, tarextumab (OMP-59R5), an anti-Notch2/3 receptor currently in phase 1 trials in patients with untreated metastatic pancreatic cancer, is showing promising therapeutic effects with the antineoplastic (chemotherapy) drugs Nab-Paclitaxel and Gemcitabine59. Examples of other Notch receptor or ligand blocking antibodies currently in clinical trials include Notch1 monoclonal antibody OMP-52M5 and the anti-DLL4 demcizumab (OMP-21M18)55.
Besides, small molecules acting as receptor or ligand decoys were also developed. A recent report by Kangsamaksin and collaborators10provided experimental evidence for the potential of decoy molecules for silencing the Notch receptors and ligands specifically60. These authors developed Notch decoys N1-13 and N1-24, interfering with Dll-Notch and Jagged-Notch interactions, respectively. The decoy molecules potently disrupted tumor growth and promoted normal endothelial sprouting in tumor microenvironment by reducing angiogenic sprouting, vessel perfusion and pericyte coverage, and other pro-angiogenic processes54. Besides the possibility to improve the therapeutic outcome, Notch receptor or ligand-specific targeting may also decrease the severity or abrogate adverse gastrointestinal effects observed with the unspecific targeting of Notch signaling.
Antimetastatic potential of Wnt and Hedgehog targeting
Wnt targeting
Wnt signaling and cancer
The Wnt family of signaling molecules encompasses a number of cysteine-rich glycosylated secreted ligands that bind to the extracellular domain of frizzled family of receptors. Wnt binding triggers a signaling cascade resulting in the activation of genes involved in stem cell maintenance, cell survival, proliferation, motility, migration, and fate determination during the development. Wnt aberrant overexpression can activate the pathogenic developmental-like Wnt signaling activity in transforming cells, favoring stemness and chemotherapy resistance61-63.
The canonical Wnt/β-catenin pathway is the most studied Wnt signaling pathway. In the canonical pathway, Wnt binding to frizzled-7 leads to the disaggregation of β-catenin destruction complex made of the tumor suppressor adenomatous polyposis coli (APC), the serine/threonine protein kinase glycogen synthase kinase 3, and casein kinase. Canonical Wnt/β-catenin pathway requires the co-receptors LRP5 and LRP6. The disaggregation of β-catenin destructioncomplex results in the inhibition of β-catenin destruction, and conversely, in the cytoplasmic accumulation of βcatenin. When in sufficiently high levels, a fraction of cytoplasmic β-catenin migrates to the nucleus where it interacts with a member of the TCF/LEF-1 family of transcription factors. Consequently, these transcription factors activate gene encoding for proteins involved in EMT, survival, angiogenesis motility, and invasion, such as c-Myc, Jagged1, VEGF, CCL2, snail, slug, vimentin, and metalloproteinases62-67.
Two other Wnt signaling pathways, i.e., the non-canonical Wnt pathways, namely, the planar cell polarity and Wnt/calcium pathway, were also described. The Wnt noncanonical pathway does not require the co-receptors LRP5/6. The non-canonical planar polarity pathway activates small Rho GTPases, such as RAC1, Rho, and Cdc42, which control actin dynamic through Rho kinase, the actin-binding protein cofilin, and the MAPK kinase JNK68-71.
The Wnt/calcium pathway, which is initiated by non-canonical Wnt ligands, such as Wnt5a, activates phosphoinositide phospholipase C (PLC) and phosphodiesterase 6 (PDE6). The PLC-mediated cleavage of the plasma membrane phospholipid phosphatidylinositol 4,5-bisphosphate releases inositol trisphosphate and diacylglycerol. The resulting signaling cascades induce the release of mitochondrial Ca2+in the cytoplasm, with subsequent activation of Ca2+-dependent enzymes, such as Ca2+/ calmodulin-dependent kinase II, protein kinase C (PKC), and the serine/threonine protein phosphatase calcineurin68-71.
Role of Wnt signaling in metastasis and therapeutic potential
Evidence sustaining a role for aberrant Wnt pathway activation in metastasis was provided by reports of the capability of Wnt antagonism to suppress tumorigenesis and metastasis partly (i) by enhancing the expression of epithelial marker, such as E-cadherin and keratin 8/18 (preventing EMT) and (ii) by decreasing the expression of pro-metastatic factors, such as slug, twist, snail, and metalloproteinases8-10. The aberrant accumulation of β-catenin in the cytoplasm observed in colorectal tumors and many other solid cancers results from tumor-promoting epigenetic events, such as DNA demethylation on a CTNNB1 gene promoter that activates β-catenin synthesis, and DNA hypermethylation on an APC gene promoter that silence this gene, resulting in the decrease of cellular levels of β-catenin destruction complex member APC70,71. Similarly, the absence of degrading the βcatenin observed in many cancers may also result from the silencing mutations of APC gene, whereas the activating mutations of CTNNB1 gene may also increase the β-catenin cellular levels72,73. The genetic and epigenetic suppression of the other members of the destruction complex was also reported74,75. Furthermore, a fraction of β-catenin is bound to the E-cadherin in the cytoskeleton, thus the EMT-associated loss of E-cadherin results in the release of βcatenin76. The failure of misshaped β-catenin (product of mutations) to bind to E-cadherin also increases the β-catenin cytoplasmic levels.
The canonical Wnt3 signaling consistently promoted a partial EMT-like transition with increased N-cadherin, twist, slug, and decreased E-cadherin in the trastuzumab-resistant breast tumor cells, and the Wnt3 knockdown by siRNA decreased the expression of EMT triggering factor EGFR in these malignant cells77. In addition, activating the Wnt /βcatenin signaling correlates with EMT and the potency of proliferation and invasiveness in prostate cancer cells63,78. The nuclear localization of β-catenin is a molecular marker of EMT in colon cancer79. The inhibition of Wnt signaling with β-catenin shRNA reversed HIF-1α-induced EMT in human prostate cancer63,78. Moreover, Wnt/β-catenin may promote EMT via direct transactivation of EMT transcription factors, such as ZEB131.
The non-canonical Wnt signaling pathways also played pivotal roles in cancer metastasis. For example, the wellknown activator of non-canonical Wnt pathway Wnt5a increased not only the metastasis in a PKC- and a Stat3-dependent manner in melanoma but also the expression of melanoma immunogens and severity markers GP100, MART-1, and tyrosinase80,81.
Wnt5a is upregulated in epithelial tumors, where it promotes EMT and migration in a β-catenin-independent manner82-89. The treatment of these tumor cells with Wnt5a resulted in an upregulation of EMT stimulating factors, including vimentin, Snail1, and slug82. In addition, Gujral and collaborators104reported an upregulation of Wnt5 receptor Frizzled2 in metastatic liver, lung, colon, and breast cancer cell lines and in high-grade tumors. Such expression correlated with the overexpression of EMT markers. In the same study, pharmacological and genetic perturbation analyses suggested that Wnt5a/Frizzled2 signaling induces EMT and cell migration via a previously unrecognized Fyn/Stat3-dependent non-canonical pathway.
Besides, considering the involvement of planar polarity pathway in regulating actin cytoskeleton mediated via its capability to activate the Rho GTPases (Section 2.3.1)68-70, a role for this non-canonical Wnt pathway in the cytoskeleton reorganization supporting motility, migration, and invasioncan be predicted. The experimental evidence suggests the involvement of this pathway in cellular motility and the invasive capacity of metastasis-promoting cells in various cancers, including pancreatic ductal adenocarcinoma and breast cancer85,86. Moreover, Frizzled receptors that can trigger canonical and non-canonical signaling, such as Frizzled2 and Frizzled7, reported the capability to interact with Rho-A and other small GTPases during metastatic cascade. For example, Wnt3a/Frizzled2 signaling stimulates cell migration and invasion in a Rho-A-dependent manner in multiple myeloma83,84, and Wnt5a was reported to promote cell migration in breast cancer through the Dishevelled2/Daam1/Rho-A axis89.
The canonical Wnt signaling pathway inhibitors, such as the CBP/β-catenin antagonist PRI-724, have been developed. The pivotal roles of this signaling pathway in maintaining the stemness of tissue stem cells and in regulating the tissue homeostasis and cell fate along the lifespan raise concerns for safety of Wnt-based anticancer strategies despite the potential of Wnt signaling targeting for cancer stem cell elimination90,91and metastasis suppression, discouraging their development. Preclinical studies91and phase 1 clinical trials92suggested that PRI-724 is relatively well-tolerated, and the drug is currently in phase 2 trials combined with chemotherapy drugs and the angiogenesis inhibitor bevacizumab (NIH trial IDs 3C-13-3, NCI-2015-00436, and NCT02413853). Experimental evidence suggests differences in Wnt signaling mediating stemness in malignant and nonmalignant cells93. The characterization of such differences may open new avenues for safe targeting of Wnt signaling.
Hedgehog, metastatic cascade, and emerging therapies
Hedgehog family
The vertebrate Hedgehog family of lipid-modified proteins encompasses Desert Hedgehog, Indian Hedgehog, and Shh. Shh is the most studied Hedgehog family member. Hedgehog proteins are the ligand of 12-pass transmembrane protein receptors, namely, patched. In the absence of interactions with ligands, patched receptors catalytically maintain the 7-pass transmembrane protein smoothened (Smo) in an inactive state. The inhibitory activity of patched is abrogated upon ligand binding, and Smo activates the transcription factors of Gli zinc-finger protein family in the cytoplasm. Then, Gli proteins migrate to the nucleus where they transactivate or repress various genes94,95.
Hedgehog played pivotal roles in cancer development96and in metastatic cancer. For example, the loss and gain and pharmacological inhibitions of function experiments suggested that Hedgehog signaling is required along the metastatic cascade97-101. Examples include studies showing (i) that high Hedgehog-Gli signatures coincide with the development of metastases in colon cancer98and (ii) that Gli1 transcription factor activity links with tumor aggressiveness in papillary thyroid cancer97. In addition, Hedgehog promotes EMT in various solid tumors, including pancreatic, colon, and breast cancers99. Gli proteins are involved in the TGF-β mediated EMT in hepatocellular carcinoma, marked by an increased expression of Snail1101. The Gli1 knockdown resulted in decreased hepatocellular carcinoma migration and invasion101. Shh enhanced the gastric cancer cell motility and invasiveness, whereas no increase was observed in cells treated with Shh signaling inhibitor cyclopamine-KAAD or anti-Shh monoclonal antibodies100. Moreover, Yoo and collaborators105demonstrated that Shh signaling promotes gastric cancer metastasis at least partly through the activation of PI3K/Akt signaling-mediated matrix metallopeptidase 9 and EMT.
Hedgehog-based therapeutic strategies
The Smo inhibitors are the largest group of Hedgehog signaling inhibitors. The naturally occurring alkaloid cyclopamine is a typical example. Cyclopamine inhibits the growth and invasiveness of a tumor cell in vitro and in vivo103-105. However, the translation to cancer treatment has been challenging due to the poor stability and solubility of cyclopamine and also due to its severe adverse effects resulting from the activity of the products of its metabolism101,106. Soluble and stable derivatives of cyclopamine were developed, including (i) the emerging prodrug cyclopamine glucuronide tested in glioblastoma multiform models107and (ii) vismodegib (GDC-0449), currently in phase 2 clinical trials in patients with metastatic pancreatic adenocarcinoma108, advanced primary tumors and metastatic basal cell carcinoma109, and pediatric brain tumors110. Other Smo inhibitors, currently in clinical trials for refractory and metastatic cancer treatment, include IPI-926111, LDE225112, BMS-833923113, and PF-0449913114. Most of these prodrugs and drugs have an acceptable safety profile with only low grade or relatively minor adverse effects with promising anticancer and antimetastatic effects. Moreover, Gli inhibitors, another class of Hedgehog inhibitors, include prodrugs, such as Gant-58 and Gant-61, which displayed a strong antimetastatic potential in preclinical studies100-123.
A number of blocking antibodies targeting Hedgehog signaling are available, while only a few inhibitors of Hedgehog ligands were developed. The monoclonal antibody5E1, one of the most studied Hedgehog-targeting antibodies, antagonizes the three Hedgehog ligands. Studies in nude mice revealed 5E1 capable of inhibiting the metastasis of pancreatic cancer118. The Hedgehog ligand inhibitor robotnikinin was discovered by Stanton and collaborators102using approaches for small molecule microarrays and diversity-oriented synthesis. This small molecule mediates extracellular Shh inhibition by binding to this protein, preventing interactions with patched receptors. Data on robotnikinin effects on metastatic processes are lacking.
Conclusions
Targeted therapies based on emerging master developmental pathways represent promising approaches for metastasis prevention and cancer stem cell elimination. Such therapeutic potential is due to the tumorigenesis-mediated hijacking of EMT, cell motility, migration, stemness, angiogenesis, and other key phenomena determining cell fate controlled by developmental pathways. These phenomena are pivotal for all steps of solid tumor initiation, maintenance, and development. Various classes of Notch signaling inhibitors are currently in clinical trials. The development of therapeutics targeting specifically the Notch signaling components critical for malignancy pathological phenotype is expected to improve the therapeutic outcome and decrease adverse effects. Wnt signaling targeting, in which safety is a serious concern, is surprisingly well-tolerated in clinical trials and is expected to be a game changer for cancer stem cell elimination and metastasis prevention. Unlike Notch and Wnt pathways, translating the Shh signaling blockade to cancer treatment has been challenging because of severe adverse effects and poor therapeutic potential of various monoclonal antibodies and naturally occurring Smo inhibitors. Further mechanistic studies of the involvement of developmental pathways in refractory and metastatic cancers may further improve the targeting efficiency and the therapeutic outcome.
Conflict of interest statement
No potential conflicts of interest are disclosed.
1.Kalluri R. EMT: when epithelial cells decide to become mesenchymal-like cells. J Clin Invest. 2009; 119: 1417-9.
2.Lenz HJ, Kahn M. Safely targeting cancer stem cells via selective catenin coactivator antagonism. Cancer Sci. 2014; 105: 1087-92.
3.Fu JS, Rodova M, Roy SK, Sharma J, Singh KP, Srivastava RK, et al. GANT-61 inhibits pancreatic cancer stem cell growth in vitro and in NOD/SCID/IL2R gamma null mice xenograft. Cancer Lett. 2013; 330: 22-32.
4.Yoo YA, Kang MH, Lee HJ, Kim BH, Park JK, Kim HK, et al. Sonic hedgehog pathway promotes metastasis and lymphangiogenesis via activation of Akt, EMT, and MMP-9 pathway in gastric cancer. Cancer Res. 2011; 71: 7061-70.
5.Clevers H. Wnt/β-catenin signaling in development and disease. Cell. 2006; 127: 469-80.
6.Wang N, Wang ZY, Wang Y, Xie XM, Shen JG, Peng C, et al. Dietary compound isoliquiritigenin prevents mammary carcinogenesis by inhibiting breast cancer stem cells through WIF1 demethylation. Oncotarget. 2015; 6: 9854-76.
7.Kwon OJ, Valdez JM, Zhang L, Zhang B, Wei X, Su Q, et al. Increased Notch signalling inhibits anoikis and stimulates proliferation of prostate luminal epithelial cells. Nat Commun. 2014; 5: 4416
8.Marchenko GN, Marchenko ND, Leng J, Strongin AY. Promoter characterization of the novel human matrix metalloproteinase-26 gene: regulation by the T-cell factor-4 implies specific expression of the gene in cancer cells of epithelial origin. Biochem J. 2002; 363: 253-62.
9.Rathinam R, Berrier A, Alahari SK. Role of Rho GTPases and their regulators in cancer progression. Front Biosci. 2011; 16: 2561-71.
10.Kangsamaksin T, Murtomaki A, Kofler NM, Cuervo H, Chaudhri RA, Tattersall IW, et al. NOTCH decoys that selectively block DLL/NOTCH or JAG/NOTCH disrupt angiogenesis by unique mechanisms to inhibit tumor growth. Cancer Discov. 2015; 5: 182-97.
11.Chiarugi P, Giannoni E. Anoikis: a necessary death program for anchorage-dependent cells. Biochem Pharmacol. 2008; 76: 1352-64.
12.Brabletz T, Jung A, Dag S, Hlubek F, Kirchner T. β-catenin regulates the expression of the matrix metalloproteinase-7 in human colorectal cancer. Am J Pathol. 1999; 155: 1033-8.
13.Justilien V, Fields AP. Molecular pathways: novel approaches for improved therapeutic targeting of Hedgehog signaling in cancer stem cells. Clin Cancer Res. 2015; 21: 505-13.
14.Bhavsar PJ, Infante E, Khwaja A, Ridley AJ. Analysis of Rho GTPase expression in T-ALL identifies RhoU as a target for Notch involved in T-ALL cell migration. Oncogene. 2013; 32: 198-208.
15.Przybylski M. A review of the current research on the role of bFGF and VEGF in angiogenesis. J Wound Care. 2009; 18: 516-9.
16.Ford CE, Punnia-Moorthy G, Henry CE, Llamosas E, Nixdorf S, Olivier J, et al. The non-canonical Wnt ligand, Wnt5a, is upregulated and associated with epithelial to mesenchymal transition in epithelial ovarian cancer. Gynecol Oncol. 2014; 134: 338-45.
17.Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. 2003; 9: 677-84.
18.Shih IM, Wang TL. Notch signaling, γ-secretase inhibitors, and cancer therapy. Cancer Res. 2007; 67: 1879-82.
19.Arwert EN, Hoste E, Watt FM. Epithelial stem cells, wound healing and cancer. Nat Rev Cancer. 2012; 12: 170-80.
20.Varnat F, Duquet A, Malerba M, Zbinden M, Mas C, Gervaz P, et al. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol Med. 2009; 1: 338-51.
21.Xu Y, An Y, Wang X, Zha W, Li X. Inhibition of the Hedgehog pathway induces autophagy in pancreatic ductal adenocarcinoma cells. Oncol Rep. 2014; 31: 707-12.
22.Li H, Da LJ, Fan WD, Long XH, Zhang XQ. Transcription factor glioma-associated oncogene homolog 1 is required for transforming growth factor-β1-induced epithelial-mesenchymal transition of non-small cell lung cancer cells. Mol Med Rep. 2015; 11: 3259-68.
23.Qi J, Yu Y, Akilli Öztürk Ö, Holland JD, Besser D, Fritzmann J, et al. New Wnt/β-catenin target genes promote experimental metastasis and migration of colorectal cancer cells through different signals. Gut. 2016; 65: 1690-701.
24.Kahn M. Can we safely target the WNT pathway? Nat Rev Drug Discov. 2014; 13: 513-32.
25.Kamga PT, Kamdje AHN. Signaling pathways in leukemia: any role for medicinal plants in leukemia therapy. J Dis Medi Plants. 2015; 1: 76-9.
26.Garg M. Epithelial-mesenchymal transition - activating transcription factors - multifunctional regulators in cancer. World J Stem Cells. 2013; 5: 188-95.
27.Alizadeh AM, Shiri S, Farsinejad S. Metastasis review: from bench to bedside. Tumour Biol. 2014; 35: 8483-523.
28.Nagaraj AB, Joseph P, Kovalenko O, Singh S, Armstrong A, Redline R, et al. Critical role of Wnt/β-catenin signaling in driving epithelial ovarian cancer platinum resistance. Oncotarget. 2015; 6: 23720-34.
29.Qiang YW, Walsh K, Yao L, Kedei N, Blumberg PM, Rubin JS, et al. Wnts induce migration and invasion of myeloma plasma cells. Blood. 2005; 106: 1786-93.
30.de Vries TJ, Smeets M, de Graaf R, Hou-Jensen K, Bröcker EB, Renard N, et al. Expression of gp100, MART-1, tyrosinase, and S100 in paraffin-embedded primary melanomas and locoregional, lymph node, and visceral metastases: implications for diagnosis and immunotherapy. A study conducted by the EORTC Melanoma Cooperative Group. J Pathol. 2001; 193: 13-20.
31.Kumamoto K, Ishida H, Ohsawa T, Ishibashi K, Ushiama M, Yoshida T, et al. Germline and somatic mutations of the APC gene in papillary thyroid carcinoma associated with familial adenomatous polyposis: Analysis of three cases and a review of the literature. Oncol Lett. 2015; 10: 2239-43.
32.Lin CH, Guo Y, Ghaffar S, McQueen P, Pourmorady J, Christ A, et al. Dkk-3, a secreted wnt antagonist, suppresses tumorigenic potential and pulmonary metastasis in osteosarcoma. Sarcoma. 2013; 2013: 147541
33.Robinson GW, Orr BA, Wu G, Gururangan S, Lin T, Qaddoumi I, et al. Vismodegib exerts targeted efficacy against recurrent sonic hedgehog-subgroup medulloblastoma: results from phase II pediatric brain tumor consortium studies PBTC-025B and PBTC-032. J Clin Oncol. 2015; 33: 2646-54.
34.O'Reilly E, Smith L, Bendell J, Strickler J, Zalupski M, Gluck W, et al. Final results of phase ib of anticancer stem cell antibody tarextumab (OMP-59R5, TRXT, anti-Notch 2/3) in combination with nab-paclitaxel and gemcitabine (Nab-P+Gem) in patients (pts) with untreated metastatic pancreatic cancer (mPC). In: 2015 Gastrointestinal Cancers Symposium. Translational Research, 2014. pp. 233-4.
35.Margadant C, Sonnenberg A. Integrin-TGF-β crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 2010; 11: 97-105.
36.Komiya Y, Habas R. Wnt signal transduction pathways. Organogenesis. 2008; 4: 68-75.
37.Xu XF, Su B, Xie CG, Wei SM, Zhou YQ, Liu H, et al. Sonic hedgehog-Gli1 signaling pathway regulates the epithelial mesenchymal transition (EMT) by mediating a new target gene, S100A4, in pancreatic cancer cells. PLoS One. 2014; 9: e96441
38.Shao S, Zhao XA, Zhang XJ, Luo MN, Zuo XX, Huang SK, et al. Notch1 signaling regulates the epithelial-mesenchymal transition and invasion of breast cancer in a Slug-dependent manner. Mol Cancer. 2015; 14: 28
39.Fei H, Ke P, Wang N, Shen H, Huang J, Tan J, et al. An evaluation comparing Californium252 neutron brachytherapy with neoadjuvant intra-arterial embolism chemotherapy assisted surgery effect for treating advanced cervical carcinoma patients. Eur J Gynaecol Oncol. 2015; 36: 442-6.
40.Dissanayake SK, Olkhanud PB, O'Connell MP, Carter A, French AD, Camilli TC, et al. Wnt5A regulates expression of tumorassociated antigens in melanoma via changes in signal transducers and activators of transcription 3 phosphorylation. Cancer Res. 2008; 68: 10205-14.
41.López-Guerra M, Xargay-Torrent S, Rosich L, Montraveta A, Roldán J, Matas-Céspedes A, et al. The γ-secretase inhibitor PF-03084014 combined with fludarabine antagonizes migration, invasion and angiogenesis in NOTCH1-mutated CLL cells. Leukemia. 2015; 29: 96-106.
42.Sher I, Adham SA, Petrik J, Coomber BL. Autocrine VEGFA/KDR loop protects epithelial ovarian carcinoma cells from anoikis. Int J Cancer. 2009; 124: 553-61.
43.Wang SS, Jiang J, Liang XH, Tang YL. Links between cancer stem cells and epithelial-mesenchymal transition. Onco Targets Ther. 2015; 8: 2973-80.
44.Wei SC, Yang J. Forcing through tumor metastasis: the interplay between tissue rigidity and epithelial-mesenchymal transition. Trends Cell Biol. 2016; 26: 111-20.
45.Ng JMY, Curran T. The Hedgehog's tale: developing strategies for targeting cancer. Nat Rev Cancer. 2011; 11: 493-501.
46.Seke Etet PF, Vecchio L, Nwabo Kamdje AH. Interactions between bone marrow stromal microenvironment and B-chronic lymphocytic leukemia cells: any role for Notch, Wnt and Hh signaling pathways? Cell Signal. 2012; 24: 1433-43.
47.Richard TS, Nwabo Kamdje AH, Mukhtar F. Medicinal plants inbreast cancer therapy. J Dis Med Plants. 2015; 1: 19-23.
48.Díaz VM, de Herreros AG. F-box proteins: Keeping the epithelialto-mesenchymal transition (EMT) in check. Semin Cancer Biol. 2016; 36: 71-9.
49.Echelard Y, Epstein DJ, St-Jacques B, Shen LY, Mohler J, McMahon JA, et al. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. 1993; 75: 1417-30.
50.Geiger TR, Peeper DS. Metastasis mechanisms. Biochim Biophys Acta. 2009; 1796: 293-308.
51.Xu ZH, Liu D, Fan CF, Luan L, Zhang XP, Wang EH. DIXDC1 increases the invasion and migration ability of non-small-cell lung cancer cells via the PI3K-AKT/AP-1 pathway. Mol Carcinog. 2014; 53: 917-925.
52.Su ZY, Yang ZZ, Xu YQ, Chen YB, Yu Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol Cancer. 2015; 14: 48
53.Pérez-Moreno MA, Locascio A, Rodrigo I, Dhondt G, Portillo F, Nieto MA, et al. A new role for E12/E47 in the repression of E-cadherin expression and epithelial-mesenchymal transitions. J Biol Chem. 2001; 276: 27424-31.
54.Leong KG, Niessen K, Kulic I, Raouf A, Eaves C, Pollet I, et al. Jagged1-mediated Notch activation induces epithelial-tomesenchymal transition through Slug-induced repression of E-cadherin. J Exp Med. 2007; 204: 2935-48.
55.Sekulic A, Migden MR, Lewis K, Hainsworth JD, Solomon JA, Yoo S, et al. Pivotal ERIVANCE basal cell carcinoma (BCC) study: 12-month update of efficacy and safety of vismodegib in advanced BCC. J Am Acad Dermatol. 2015; 72: 1021-6.e8.
56.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014; 15: 178-96.
57.Yeo EJ, Cassetta L, Qian BZ, Lewkowich I, Li JF, Stefater III JA, et al. Myeloid WNT7b mediates the angiogenic switch and metastasis in breast cancer. Cancer Res. 2014; 74: 2962-73.
58.Cetin GO, Toylu A, Atabey N, Sercan Z, Sakizli M. Downregulation of VANGL1 inhibits cellular invasion rather than cell motility in hepatocellular carcinoma cells without stimulation. Genet Test Mol Biomarkers. 2015; 19: 283-7.
59.Ma HK, Li HQ, Zhang X. Cyclopamine, a naturally occurring alkaloid, and its analogues may find wide applications in cancer therapy. Curr Top Med Chem. 2013; 13: 2208-15.
60.Lee SM, Moon J, Redman BG, Chidiac T, Flaherty LE, Zha YY, et al. Phase 2 study of RO4929097, a gamma-secretase inhibitor, in metastatic melanoma: SWOG 0933. Cancer. 2015; 121: 432-40.
61.Vazquez-Martin A, Cufí S, Oliveras-Ferraros C, Torres-Garcia VZ, Corominas-Faja B, Cuyàs E, et al. IGF-1R/epithelial-tomesenchymal transition (EMT) crosstalk suppresses the erlotinibsensitizing effect of EGFR exon 19 deletion mutations. Sci Rep. 2013; 3: 2560
62.Martin SS, Vuori K. Regulation of Bcl-2 proteins during anoikis and amorphosis. Biochim Biophys Acta. 2004; 1692: 145-57.
63.Gómez-Orte E, Sáenz-Narciso B, Moreno S, Cabello J. Multiple functions of the noncanonical Wnt pathway. Trends Genet. 2013; 29: 545-53.
64.Citi S, Guerrera D, Spadaro D, Shah J. Epithelial junctions and Rho family GTPases: the zonular signalosome. Small GTPases. 2014; 5: e973760
65.Jiang YG, Luo Y, He DL, Li X, Zhang LL, Peng T, et al. Role of Wnt/β-catenin signaling pathway in epithelial-mesenchymal transition of human prostate cancer induced by hypoxia-inducible factor-1α. Int J Urol. 2007; 14: 1034-9.
66.Wu YY, Ginther C, Kim J, Mosher N, Chung S, Slamon D, et al. Expression of Wnt3 activates Wnt/β-catenin pathway and promotes EMT-like phenotype in trastuzumab-resistant HER2-overexpressing breast cancer cells. Mol Cancer Res. 2012; 10: 1597-606.
67.Fodouop SPC, Simo RT, Amvene JM, Talla T, Seke Etet PF, Takam P, et al. Bioactivity and therapeutic potential of plant extracts in cancer and infectious diseases. J Dis Med Plants. 2015; 1: 8-18.
68.Zhu YC, Tian YH, Du J, Hu ZZ, Yang L, Liu JJ, et al. Dvl2-dependent activation of Daam1 and RhoA regulates Wnt5ainduced breast cancer cell migration. PLoS One. 2012; 7: e37823
69.Villanueva MT. Angiogenesis: a sudden rush of blood to the tumour. Nat Rev Cancer. 2015; 15: 135
70.Gulhati P, Bowen KA, Liu JY, Stevens PD, Rychahou PG, Chen M, et al. mTORC1 and mTORC2 regulate EMT, motility, and metastasis of colorectal cancer via RhoA and Rac1 signaling pathways. Cancer Res. 2011; 71: 3246-56.
71.Bailey JM, Mohr AM, Hollingsworth MA. Sonic hedgehog paracrine signaling regulates metastasis and lymphangiogenesis in pancreatic cancer. Oncogene. 2009; 28: 3513-25.
72.Lee J, Jeong S, Lee CR, Ku CR, Kang SW, Jeong JJ, et al. GLI1 transcription factor affects tumor aggressiveness in patients with papillary thyroid cancers. Medicine. 2015; 94: e998
73.Barnes EA, Kenerson HL, Jiang XY, Yeung RS. Tuberin regulates E-cadherin localization: implications in epithelial-mesenchymal transition. Am J Pathol. 2010; 177: 1765-78.
74.Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004; 20: 781-810.
75.Guo SC, Liu ML, Gonzalez-Perez RR. Role of Notch and its oncogenic signaling crosstalk in breast cancer. Biochim Biophys Acta. 2011; 1815: 197-213.
76.Hatakeyama J, Wald JH, Printsev I, Ho HYH, Carraway III KL. Vangl1 and Vangl2: planar cell polarity components with a developing role in cancer. Endocr Relat Cancer. 2014; 21: R345-56.
77.Chung MT, Lai HC, Sytwu HK, Yan MD, Shih YL, Chang CC, et al. SFRP1 and SFRP2 suppress the transformation and invasion abilities of cervical cancer cells through Wnt signal pathway. Gynecol Oncol. 2009; 112: 646-53.
78.Martin-Villar E, Megías D, Castel S, Yurrita MM, Vilaró S, Quintanilla M. Podoplanin binds ERM proteins to activate RhoA and promote epithelial-mesenchymal transition. J Cell Sci. 2006; 119: 4541-53.
79.Hassan WA, Yoshida R, Kudoh S, Hasegawa K, Niimori-Kita K,Ito T. Notch1 controls cell invasion and metastasis in small cell lung carcinoma cell lines. Lung Cancer. 2014; 86: 304-10.
80.Kim S, Kang M, Lee S, Bae S, Han S, Jang JY, et al. Identifying molecular subtypes related to clinicopathologic factors in pancreatic cancer. Biomed Eng Online. 2014; 13 Suppl 2: S5
81.Ko AH, LoConte N, Tempero MA, Walker EJ, Kate KR, Lewis S, et al. A phase I study of FOLFIRINOX Plus IPI-926, a hedgehog pathway inhibitor, for advanced pancreatic adenocarcinoma. Pancreas. 2016; 45: 370-5.
82.Migden MR, Guminski A, Gutzmer R, Dirix L, Lewis KD, Combemale P, et al. Treatment with two different doses of sonidegib in patients with locally advanced or metastatic basal cell carcinoma (BOLT): a multicentre, randomised, double-blind phase 2 trial. Lancet Oncol. 2015; 16: 716-28.
83.Sanchez-Tillo E, de Barrios O, Siles L, Cuatrecasas M, Castells A, Postigo A. beta-catenin/TCF4 complex induces the epithelial-tomesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proc Natl Acad Sci U S A. 2011; 108: 19204-9.
84.Schödel J, Grampp S, Maher ER, Moch H, Ratcliffe PJ, Russo P, et al. Hypoxia, hypoxia-inducible transcription factors, and renal cancer. Eur Urol. 2016; 69: 646-57.
85.Johnson RW, Schipani E, Giaccia AJ. HIF targets in bone remodeling and metastatic disease. Pharmacol Ther. 2015; 150: 169-77.
86.Warzecha J, Dinges D, Kaszap B, Henrich D, Marzi I, Seebach C. Effect of the Hedgehog-inhibitor cyclopamine on mice with osteosarcoma pulmonary metastases. Int J Mol Med. 2012; 29: 423-7.
87.Hayes JD, Chowdhry S, Dinkova-Kostova AT, Sutherland C. Dual regulation of transcription factor Nrf2 by Keap1 and by the combined actions of β-TrCP and GSK-3. Biochem Soc Trans. 2015; 43: 611-20.
88.Zhu HH, Zhu XY, Zhou MH, Cheng GY, Lou WH. Effect of WNT5A on epithelial-mesenchymal transition and its correlation with tumor invasion and metastasis in nasopharyngeal carcinoma. Asian Pac J Trop Med. 2014; 7: 488-91.
89.Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelialmesenchymal transition: new insights in signaling, development, and disease. J Cell Biol. 2006; 172: 973-81.
90.Bensalma S, Chadeneau C, Legigan T, Renoux B, Gaillard A, de Boisvilliers M, et al. Evaluation of cytotoxic properties of a cyclopamine glucuronide prodrug in rat glioblastoma cells and tumors. J Mol Neurosci. 2015; 55: 51-61.
91.Wang YQ. Wnt/Planar cell polarity signaling: a new paradigm for cancer therapy. Mol Cancer Ther. 2009; 8: 2103-2109.
92.Kumar S, Park SH, Cieply B, Schupp J, Killiam E, Zhang F, et al. A pathway for the control of anoikis sensitivity by E-cadherin and epithelial-to-mesenchymal transition. Mol Cell Biol. 2011; 31: 4036-51.
93.Zheng X, Vittar NB, Gai XH, Fernandez-Barrena MG, Moser CD, Hu CL, et al. The transcription factor GLI1 mediates TGFβ1 driven EMT in hepatocellular carcinoma via a SNAI1-dependent mechanism. PLoS One. 2012; 7: e49581
94.Lauth M, Bergstrom A, Shimokawa T, Toftgard R. Inhibition of GLI-mediated transcription and tumor cell growth by smallmolecule antagonists. Proc Natl Acad Sci U S A. 2007; 104: 8455-60.
95.Yu M, Ting DT, Stott SL, Wittner BS, Ozsolak F, Paul S, et al. RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature. 2012; 487: 510-3.
96.Kandala PK, Srivastava SK. Diindolylmethane-mediated Gli1 protein suppression induces anoikis in ovarian cancer cells in vitro and blocks tumor formation ability in vivo. J Biol Chem. 2012; 287: 28745-54.
97.Zhao JH, Luo Y, Jiang YG, He DL, Wu CT. Knockdown of β-Catenin through shRNA cause a reversal of EMT and metastatic phenotypes induced by HIF-1α. Cancer Invest. 2011; 29: 377-82.
98.Stoletov K, Lewis JD. Invadopodia: a new therapeutic target to block cancer metastasis. Expert Rev Anticancer Ther. 2015; 15: 733-5.
99.Ozawa M, Kobayashi W. Reversibility of the Snail-induced epithelial-mesenchymal transition revealed by the Cre-loxP system. Biochem Biophys Res Commun. 2015; 458: 608-13.
100.Pajic M, Herrmann D, Vennin C, Conway JRW, Chin VT, Johnsson AK, et al. The dynamics of Rho GTPase signaling and implications for targeting cancer and the tumor microenvironment. Small GTPases. 2015; 6: 123-33.
101.Andersson ER, Lendahl U. Therapeutic modulation of Notch signalling--are we there yet? Nat Rev Drug Discov. 2014; 13: 357-78.
102.Stanton BZ, Peng LF, Maloof N, Nakai K, Wang X, Duffner JL, et al. A small molecule that binds Hedgehog and blocks its signaling in human cells. Nat Chem Biol. 2009; 5: 154-6.
103.Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. 2015; 12: 445-64.
104.Gujral TS, Chan M, Peshkin L, Sorger PK, Kirschner MW, MacBeath G. A noncanonical Frizzled2 pathway regulates epithelial-mesenchymal transition and metastasis. Cell. 2014; 159: 844-56.
105.Yoo YA, Kang MH, Kim JS, Oh SC. Sonic hedgehog signaling promotes motility and invasiveness of gastric cancer cells through TGF-β-mediated activation of the ALK5-Smad 3 pathway. Carcinogenesis. 2008; 29: 480-90.
106.Khan AH, Bloom JS, Faridmoayer E, Smith DJ. Genetic screening reveals a link between Wnt signaling and antitubulin drugs. Pharmacogenomics J. 2016; 16: 164-72.
107.Ntziachristos P, Lim JS, Sage J, Aifantis I. From fly wings to targeted cancer therapies: a centennial for notch signaling. Cancer Cell. 2014; 25: 318-34.
108.Filipović A, Lombardo Y, Faronato M, Abrahams J, Aboagye E, Nguyen QD, et al. Anti-nicastrin monoclonal antibodies elicit pleiotropic anti-tumour pharmacological effects in invasive breast cancer cells. Breast Cancer Res Treat. 2014; 148: 455-62.
109.Nieto MA. The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol. 2002; 3: 155-66.
110.Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev. 2008; 22: 2454-72.
111.Fortin Ensign SP, Mathews IT, Symons MH, Berens ME, Tran NL. Implications of Rho GTPase signaling in glioma cell invasion and tumor progression. Front Oncol. 2013; 3: 241
112.Paoli P, Giannoni E, Chiarugi P. Anoikis molecular pathways and its role in cancer progression. Biochim Biophys Acta. 2013; 1833: 3481-98.
113.Tejeda-Muñoz N, Robles-Flores M. Glycogen synthase kinase 3 in Wnt signaling pathway and cancer. IUBMB Life. 2015; 67: 914-22.
114.van Zijl F, Krupitza G, Mikulits W. Initial steps of metastasis: cell invasion and endothelial transmigration. Mutat Res. 2011; 728: 23-34.
115.Chen WT, Lee CC, Goldstein L, Bernier S, Liu CHL, Lin CY, et al. Membrane proteases as potential diagnostic and therapeutic targets for breast malignancy. Breast Cancer Res Treat. 1994; 31: 217-26.
116.Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, et al. Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science. 1997; 275: 1787-90.
117.Tian XR, Liu ZL, Niu B, Zhang JL, Tan TK, Lee SR, et al. E-cadherin/β-catenin complex and the epithelial barrier. J Biomed Biotechnol. 2011; 2011: 567305
118.Schwarzer R, Nickel N, Godau J, Willie BM, Duda GN, Schwarzer R, et al. Notch pathway inhibition controls myeloma bone disease in the murine MOPC315.BM model. Blood Cancer J. 2014; 4: e217
119.Fan Q, Gu D, He M, Liu H, Sheng T, Xie G, et al. Tumor shrinkage by cyclopamine tartrate through inhibiting hedgehog signaling. Chin J Cancer. 2011; 30: 472-81.
120.Parmalee NL, Kitajewski J. Wnt signaling in angiogenesis. Curr Drug Targets. 2008; 9: 558-64.
121.Ricciardi M, Malpeli G, Bifari F, Bassi G, Pacelli L, Nwabo Kamdje AH, et al. Comparison of epithelial differentiation and immune regulatory properties of mesenchymal stromal cells derived from human lung and bone marrow. PLoS One. 2012; 7: e35639.
122.Qiang YW, Endo Y, Rubin JS, Rudikoff S. Wnt signaling in B-cell neoplasia. Oncogene. 2003; 22: 1536-45.
123.Qiang YW, Ye S, Chen Y, Buros AF, Edmonson R, van Rhee F, et al. MAF protein mediates innate resistance to proteasome inhibition therapy in multiple myeloma. Blood. 2016; 128: 2919-302.
124.Espinoza I, Miele L. Deadly crosstalk: Notch signaling at the intersection of EMT and cancer stem cells. Cancer Lett. 2013; 341: 41-5.
125.Krause U, Ryan DM, Clough BH, Gregory CA. An unexpected role for a Wnt-inhibitor: Dickkopf-1 triggers a novel cancer survival mechanism through modulation of aldehydedehydrogenase-1 activity. Cell Death Dis. 2014; 5: e1093
126.Seke Etet PF, Nwabo Kamdje AH, Mbo Amvene J, Aldebasi Y, Farahna M, Vecchio L. Stromal control of chronic lymphocytic leukemia cells. Res Rep in Biol. 2013; 4:23-32.
Cite this article as: Nwabo Kamdje AH, Takam Kamga P, Tagne Simo R, Vecchio L, Seke Etet PF, Muller JM, et al. Developmental pathways associated with cancer metastasis: Notch, Wnt, and Hedgehog. Cancer Biol Med. 2017; 14: 109-20. doi: 10.20892/j.issn.2095-3941.2016.0032
Armel Herve Nwabo Kamdje
E-mail: armel.nwabo@gmail.com
March 22, 2016; accepted March 27, 2017.
Available at www.cancerbiomed.org
Copyright © 2017 by Cancer Biology & Medicine
 Cancer Biology & Medicine2017年2期
Cancer Biology & Medicine2017年2期
- Cancer Biology & Medicine的其它文章
- Cellular immunity augmentation in mainstream oncologic therapy
- Mesenchymal stromal cells’ role in tumor microenvironment: involvement of signaling pathways
- Simplified microsatellite instability detection protocol provides equivalent sensitivity to robust detection strategies in Lynch syndrome patients
- Significance of stromal-1 and stromal-2 signatures and biologic prognostic model in diffuse large B-cell lymphoma
- Immunohistochemical evaluation of vitamin D receptor(VDR) expression in cutaneous melanoma tissues and four VDR gene polymorphisms
- Concomitant-chemoradiotherapy-associated oral lesions in patients with oral squamous-cell carcinoma