
抗肿瘤多肽9R-P201诱导下的肝癌HepG2细胞转录组测序分析
2017-07-12 18:05:57刘文荣丁若凡张一鸣李宇鹏李玲郭志云
生物技术通报 2017年7期
刘文荣丁若凡张一鸣李宇鹏李玲郭志云
(1. 西南交通大学生命科学与工程学院,成都 610031;2. 成都市第三人民医院病理科,成都 610031)
抗肿瘤多肽9R-P201诱导下的肝癌HepG2细胞转录组测序分析
刘文荣1丁若凡1张一鸣1李宇鹏1李玲2郭志云1
(1. 西南交通大学生命科学与工程学院,成都 610031;2. 成都市第三人民医院病理科,成都 610031)
旨在探索多肽9R-P201处理肝癌HepG2细胞后基因融合、单核苷酸多态性(Single nucleotide polymorphism,SNP)突变、可变剪接等事件,并分析差异表达基因所参与的生物学进程与信号通路,以期解析多肽9R-P201在转录组水平对肝癌细胞的调控。通过转录组测序检测9R-P201处理肝癌HepG2细胞前后基因差异表达情况,tophat-fusion软件检测基因融合,SAMTOOLS软件检测SNP位点,rMATS软件鉴定可变剪接,使用基因本体(Gene Ontology,GO)和京都基因与基因组百科全书(Kyoto encyclopedia of genes and genomes,KEGG)富集分析方法对差异表达基因进行功能富集分析。结果共检测到可变剪接事件276个、SNP位点5 557个、基因融合事件45个;同时共得到显著差异表达基因 403个,其中上调269个而下调134个,基因的功能富集分析结果显示差异表达基因显著富集细胞生长、迁移等肿瘤相关生物进程,并参与多条与癌症相关的信号通路。研究表明在9R-P201诱导HepG2细胞后,导致表达差异基因显著与肿瘤生物学进程和通路相关,并发生了大量可变剪接、SNP突变、基因融合等事件,这暗示着该多肽有望作为后续肝癌介入治疗潜在药物分子。
9R-P201;肝细胞癌;转录组测序;基因
在全世界范围内,肝细胞癌(Hepatocellular carcinoma,HCC)是最为常见的恶性肿瘤之一,死亡率高居所有肿瘤第二位[1]。目前除了肝移植、手术以及放化疗等治疗手段外,分子靶向药物在治疗肝细胞癌方面也取得了许多重要的进展[2]。目前靶向肝细胞癌药物主要包括化学药物和多肽药物,其中索拉菲尼、舒尼替尼、瓦他拉尼等分子靶向化学药物已在临床上应用[3]。除此之外,靶向肝细胞癌的多肽药物的研究也取得明显进展,多肽药物是由人工合成的或经分离纯化得到的活性多肽,先前研究发现一些小分子多肽在抑制肿瘤的发生与发展中起着重要的作用。例如,蛋氨酸脑啡肽(Methionine enkephalin,MENK)是具有增强体液免疫和提高细胞杀伤作用的内源性多肽,研究表明MENK通过精确调控树状细胞的阿片类药物受体介导的功能来发挥抗肿瘤的活性[4]。研究证实从家蚕幼虫中分离得到的抗菌肽CecropinXJ 能抑制肝癌细胞的生长并诱导凋亡,这暗示着CecropinXJ 或许可作为抗肝癌的多肽药物[5]。
本实验室前期通过噬菌体随机12肽库筛选得到了一个多肽9R-P201,实验证实9R-P201可显著抑制肝癌HepG2细胞的存活、增殖和迁移并诱导凋亡[6,7]。为了探寻多肽9R-P201在转录组水平抑制肝细胞癌的形成发展的机理,本研究利用转录组测序对9R-P201处理HepG2细胞前后的基因表达谱进行分析,筛选出了一系列差异表达基因,并证明其生物学功能。
1 材料与方法
1.1 材料
人肝癌细胞HepG2细胞株来自于四川大学生命学院,由本实验室保管;9R-P201购自上海强耀生物科技有限公司;DMEM培养基购自Gibco公司;100 U/mL青霉素和100 U/mL链霉素购自Hyclone公司;胎牛血清购自Biowest公司;Trizol Regent购自Invitrogen公司;转录组测序由北京百迈客生物科技有限公司完成。
1.2 方法
1.2.1 细胞培养 HepG2细胞用含有10% 胎牛血清(Biowest),100 IU/mL青霉素和100 mg/mL链霉素(Hyclone)的DMEM培养基(Gibco),并置于37℃、5% CO2饱和湿度的细胞培养箱中进行培养。取处于对数生长期的细胞3.0×105个接种于6孔板中,培养过夜至细胞融合度达到80 %左右,用80 μg/mL的9R-P201(上海强耀生物)作用于HepG2细胞,处理24 h后,收集细胞用于后续的RNA提取。
1.2.2 RNA提取及质量检测 收集好的9R-P201处理和未处理的HepG2细胞用Trizol试剂(Invitrogen)按试剂说明书提取细胞总RNA。1 %琼脂糖凝胶电泳和酶标仪(Biotech)鉴定RNA的纯度、浓度及完整性,合格的总RNA保存于-80℃冰箱待用。
1.2.3 RNA文库构建、测序 RNA检测合格后,用带有Oligo(dT)的磁珠进行富集并随机打断,以短片段RNA为模板,用六碱基随机引物(random hexamers)合成一链cDNA,然后加入缓冲液、dNTPs、RNase和DNA polymerase I合成二链cDNA。纯化的双链cDNA再进行末端修复、加A尾并连接测序接头,最后进行PCR富集得到链特异性cDNA文库,使用Illumina HiSeq 4000测序仪进行文库测序。
1.2.4 转录组测序数据的处理、比对和组装 测序原始测序数据通过去除带接头和低质量的reads后获得高质量的clean reads。使用Tophat2软件将clean reads比对到hg19参考基因组上,用Cufflinks软件按照hg19参考基因组进行组装[8]。
1.2.5 可变剪接鉴定、基因融合事件鉴定和SNP位点分析 使用rMATS软件进行可变剪接的鉴定,在本次转录组测序分析中我们主要对常见的5种可变剪接类型进行统计。使用tophat-fusion软件来鉴定基因融合,最后使用Circos可视化工具来绘制基因融合示意图[9]。将每个样品的测序结果与参考基因组相比,使用SAMTOOLS软件检测SNP位点,并使用ANNOVAR工具对SNP位点进行注释[10]。
1.2.6 差异表达基因及功能富集分析 采用Audic S算法进行差异表达分析,将错误发现率(False discovery rate,FDR)校正后的q< 0.05且差异倍数|log2FC|≥1的视为差异表达基因,并对筛选得到的差异表达基因进行聚类分析。利用GO数据库和KEGG数据库对差异表达的基因进行GO与信号通路富集分析,以q < 0.05为显著性阈值[11,12]。
2 结果
2.1 转录组测序与差异分析基因分析
转录组测序得到对照组和处理组总的reads数分别有46 698 580和39 332 045条,过滤得到clean reads数所占的比例分别为99.45%和97.46%。本次转录组测序共检测到18 152个基因,按照差异表达筛选标准得到差异表达基因 403个,其中上调269个下调134个,我们对筛选得到的差异表达基因分析表明9R-P201处理后基因整体呈上调趋势(图1)。
2.2 可变剪接与基因融合事件鉴定
对常见的5种可变剪接类型的数量进行了统计,结果发现在本次转录组测序中一共检测到276个可变剪接,其中外显子跳跃(Exon skipping,ES)最多有126个而内含子保留(Intron Retention,RI)最少有12个,此外3'端可变剪接(Alternative 3' splice site,A3SS)有54个、5'端可变剪接(Alternative 5' splice site,A5SS)有60个、互斥外显子(Mutually exclusive exon,MXE)有24个(图2-A)。
异常的融合基因可以引起恶性血液疾病以及肿瘤,为此进行了基因融合分析。结果共鉴定得到45个基因融合,发现基因融合事件主要发生在11号、12号和19号染色体上(图2-B)。在这45个基因融合中,FTH1、GAPDH、KRT7等基因与其它基因发生融合的频率比较高(表1)。
2.3 SNP分析
通过在对照组和处理组进行SNP分析发现在对照组和处理组中分别得到SNP位点2 180个和3 377个,其中对照组的2 180个SNP包括816个非同义突变SNP(Nonsynonymous SNP)和1 364个同义突变SNP(Synonymous SNP),处理组的3 377个SNP包括1 249个非同义突变SNP(Nonsynonymous SNP)和2 128个同义突变SNP(Synonymous SNP);同义突变SNP和非同义突变SNP中都包括6种类型SNP突变,结果(图2-C)发现C/T的突变频率最高而A/T的突变频率最低。

图1 差异表达基因分析
2.4 差异表达基因功能分析
通过GO和KEGG富集分析对403个差异表达基因进行功能注释,以q < 0.05为显著性阈值。GO富集分析一共富集上34条GO term,包括生物学过程(BP)18条、细胞成分(CC)8条以及分子功能(MF)8条,其中富集上与肿瘤相关的GO term有细胞生长(q = 5.05E-08)、细胞移动(q = 8.19 E-07)、细胞信号转导(q = 9.91 E-06)等(图3-A)。KEGG信号通路富集分析结果(图3-B)表明,差异表达基因显著地富集上肿瘤转录失调(q = 4.66 E-08)、细胞因子受体相互作用(q = 3.84 E-06)、细胞间隙连接(q = 3.40E-05)等信号通路。
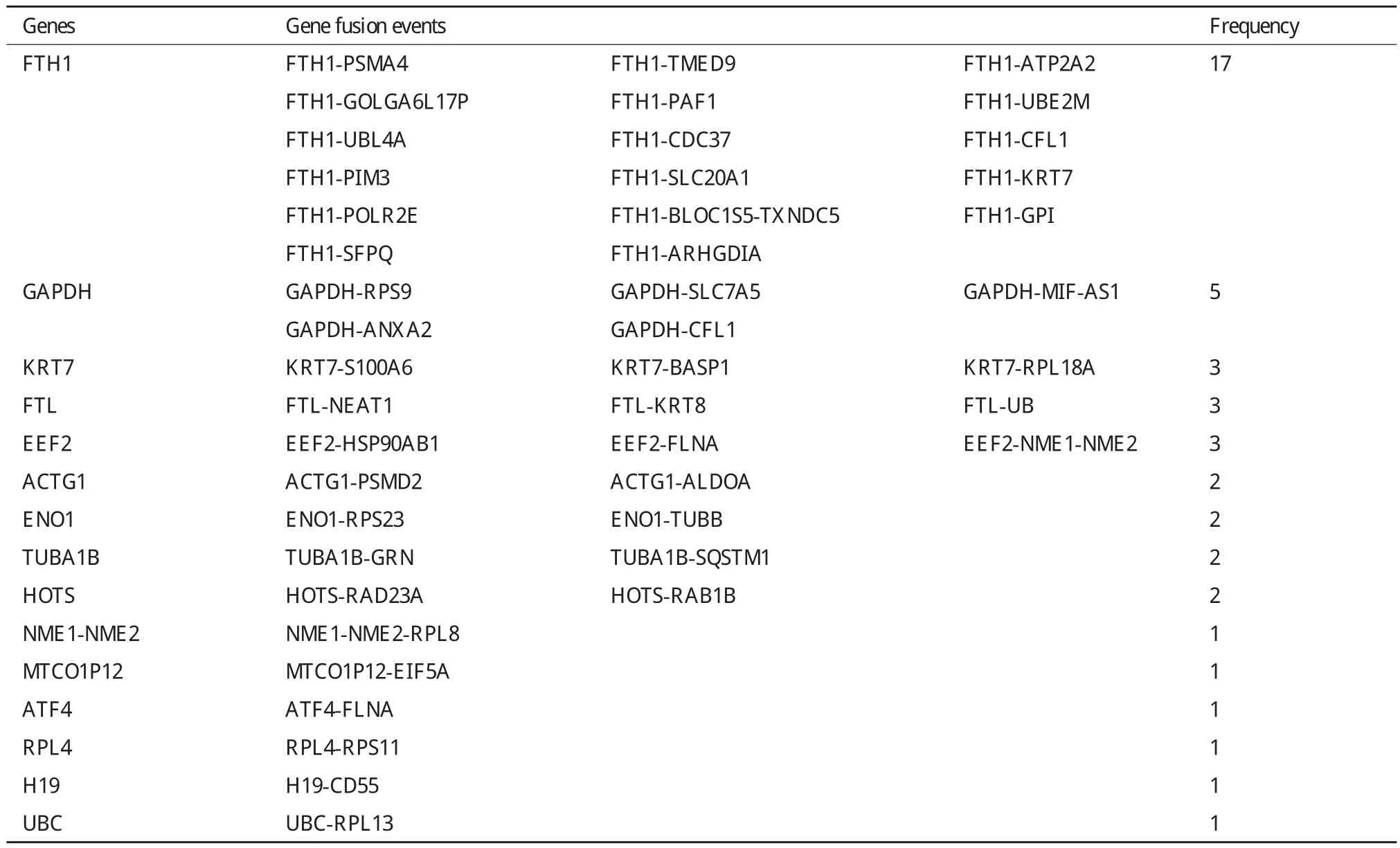
表1 基因融合事件统计
3 讨论
本次转录组测序筛选得到显著性差异表达基因有403个,其中上调的有269个而下调的有134个。GO和KEGG分析结果表明差异表达基因显著参与了与肿瘤相关的生物学进程和信号通路。对5种主要的可变剪接类型进行统计,共得到可变剪接位点276个。本研究中一共得到SNP位点5 557个。在SNP位点中,非同义突变SNP位点有2 065个而同义突变SNP位点有3 492个,同义突变所占的比例在70%左右而非同义突变占30%左右,这与文献报道的结果一致[13]。同时在SNP突变类型中,C/T和A/G突变的频率最高,这可能是因为在人类基因组中存在大量的CpG岛,同时CpG岛中的C常常是甲基化的并容易发生脱氨基而转变为T[14]。另外,我们这些获取的SNP位点还存在一定的假阳性,如肝细胞癌HepG2细胞系经过长时间的培养,其染色体经常存在非整数倍的染色体变化并且包含如删除和复制等的染色体改变,这些染色体的变化会直接影响SNP的检测结果[15]。由于RNA-seq检测的RNA在进行编辑的过程中也会导致序列变异从而可能被错误地注释为SNP,因此一些检测到的序列变异有可能是RNA编辑位点,而不是真正的SNP[16,17]。此外,由于无法保证对照组与处理组RNA样本的完全一致,多肽9R-P201处理后导致的SNP中还会存在由于对照组与处理组RNA样本不一致导致的SNP假阳性。对基因融合进行鉴定,共得到45个基因融合位点,其中FTH1基因与其它基因发生融合的次数有17次;研究发现FTH1基因在离子储存和转运起重要作用并调控细胞内离子内稳态[18]。
综上所述,这些显著差异表达的基因可能在多肽9R-P201激活的生物学功能和信号通路中起着重要的功能。同时可变剪接、SNP位点、基因融合等基因组结构的改变也暗示多肽9R-P201在这些事件上对肝癌HepG2细胞的调控影响。当然,9R-P201对于肝癌细胞的调控不仅局限于基因来发挥作用,通过发现更多的9R-P201下游调控因子以及信号通路将为基于9R-P201多肽为基础的药物开发将提供更多的支撑。

图2 可变剪接、基因融合、SNP分析结果
4 结论
在肝癌发生发展过程中涉及多个与癌症相关的生物学过程和信号通路的改变。本研究分析了肝癌HepG2细胞在9R-P201作用下基因差异表达情况、可变剪接、SNP位点和基因融合等。共得到可变剪接位点276个、SNP位点5 557个及基因融合位点45个。此外,还得到显著差异表达的基因 403个,其中上调269个,下调134个。基因功能注释结果表明差异表达基因富集上多个与肿瘤密切相关的生物学进程和信号通路,预示这些基因在9R-P201对肝癌HepG2细胞的调控过程中发挥重要的作用。
[1]Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide:sources, methods and major patterns in GLOBOCAN 2012[J]. Int J Cancer, 2015, 136(5):E359-386.
[2]Brito AF, Abrantes AM, Tralhão JG, et al. Targeting hepatocellular carcinoma:what did we discover so far?[J]. Oncology Reviews, 2016, 10(2):302.
[3]Deng GL, Zeng S, Shen H. Chemotherapy and target therapy for hepatocellular carcinoma:New advances and challenges[J]. World Journal of Hepatology, 2015, 7(5):787-798.
[4]Meng Y, Gao X, Chen W, et al. Methionine enkephalin(MENK)mounts antitumor effect via regulating dendritic cells(DCs)[J]. International Immunopharmacology, 2017, 44:61-71.
[5]Xia L, Wu Y, Ma JI, et al. The antibacterial peptide from Bombyxmori cecropinXJ induced growth arrest and apoptosis in human hepatocellular carcinoma cells[J]. Oncology Letters, 2016, 12(1):57-62.
[6]Cui J, Huang J, Guo T, et al. Expression and selection of human foxm1c binding peptides and yheir inhibitions on MCF7 cancer cells[J]. Int J Pept Res Ther, 2014, 20(4):447-456.
[7]Bi Z, Liu W, D ing R, et al. A novel peptide, 9R-P201, strongly inhibits the viability, proliferation and migration of liver cancer HepG2 cells and induces apoptosis by down-regulation of FoxM1 expression[J]. Eur J Pharmacol, 2017, 796:175-189.
[8]Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq e xperiments with TopHat and Cufflinks[J]. Nature Protocols, 2012, 7(3):562-578.
[9]Kim D, Salzberg SL. TopHat-Fusion:an algorithm for discovery of novel fusion transcripts[J]. Genome Biol, 2011, 12(8):R72.
[10]Li HD, Menon R, Omenn GS, et al. The emerging era of genomic data integration for analyzing splice isoform function[J]. Trends in Gene tics:TIG, 2014, 30(8):340-347.
[11]Thomas PD, Mi H, Lewis S. Ontology annotation:mapping genomic regions to biological function[J]. Current Opinion in Chemical Biology, 2007, 11(1):4-11.
[12]Kanehi sa M, Araki M, Goto S, et al. KEGG for linking genomes to life and the environment[J]. Nucleic Acids Res, 2008, 36:D480-484.
[13] Koberle B, Koch B, Fischer BM, et al. Single nucleotide polymorphisms i n DNA repair genes and putative cancer risk[J]. Archives of Toxicology, 2016, 90(10):2369-2388.
[14]Shastry BS. SNPs:impact on gene function and phenotype[J]. Methods in Molecular Biology, 2009, 578:3-22.
[15]Chen J, Driguneswar P, Shravan K, et al. Selecting c ell lines for SNP human identification assay development[J]. Atlas Journal of Biotechnology, 2011, 1(1):21-26, 2011.
[16]Gommans WM, Tatalias NE, Sie CP, et al. Screening of human SNP database identifies recoding sites of A-to-I RNA editing[J]. RNA, 2008, 14(10):2074-2085.
[17]Eisenberg E, Adamsky K, Cohen L, et al. Identification of RNA editing sites in the SNP database[J]. Nucleic Acids Research, 2005, 33(14):4612-4617.
[18]Guo J, Xu N, Yao Y, et al. Efficient expression of rec ombinant human heavy chain ferritin(FTH1)with modified peptides[J]. Protein Expression and Purification, 2016, 131:101-108.

图3 差异表达基因功能富集分析
(责任编辑 李楠)
Transcriptome Sequencing Analysis of Hepatocellular Carcinoma HepG2 Cells Induced by Antitumor Peptide 9R-P201
LIU Wen-rong1DING Ruo-fan1ZHANG Yi-ming1LI Yu-peng1LI Ling2GUO Zhi-yun1
(1. School of Life Science and Engineering,Southwest Jiaotong University,Chengdu 610031;2. Department of Pathology,the Third People’s Hospital of Chengdu,Chengdu 610031)
This wok aims to elucidate the regulation of 9R-P201 on hepatoma cells(HepG2)at transcriptome level by investigating the gene fusion,SNP(Single Nucleotide Polymorphism)mutation,alternative splicing and other events after 9R-P201 treating HepG2,and analyzing the biological processes and signaling pathways involved by differentially expressed genes. The expression differences of the genes before and after the 9R-P201 treating HepG2 cell line were detected by transcriptome sequencing. Meanwhile,gene fusion,SNPs,and alternative splicing were identified by tophat-fusion,SAMTOOLS software,and rMATS respectively. Functional enrichment analysis of differentially expressed genes were performed by GO(Gene Ontology)and KEGG(Kyoto Encyclopedia of Genes and Genomes). As results,276 alternative splicing events,5 557 SNP sites,and 45 gene fusion events were detected in the transcriptome sequencing. In addition,403 differentially expressed genes including 269 up-regulated and 134 down-regulated were detected. Gene GO and KEGG enrichment analysis showed that differentially expressed genes were significantly involved in the biological processes of cell growth,locomotion,as well as a number of cancer-related signaling pathways. In conclusion,the study revealed that the differentially expressed genes resulted from 9R-P201 treating HepG2 were significantly correlated with the biological processes and signaling pathways related to cancer and hepatocellular carcinoma HepG2 cell line,and a large number of alternative splicing events,SNP mutations,gene fusion events after 9R-P201 inducement occurred,suggesting this peptide may be used as a potential drug for subsequent hepatocellular carcinoma interventional therapy.
9R-P201;hepatocellular carcinoma;transcriptome sequencing;gene
10.13560/j.cnki.biotech.bull.1985.2017-0053
2017-01-26
中央高校基本科研业务费专项资金(2682016YXZT04),国家大学生创新性实验计划项目(201610613066)
刘文荣,男,硕士研究生,研究方向:非编码RNA结构与功能;E-mail:liuzimu1992@gmail.com
郭志云,男,副教授,研究方向:生物信息学;E-mail:zhiyunguo@gmail.com
猜你喜欢
新民周刊(2022年27期)2022-08-01 07:04:49
上海金属(2021年6期)2021-12-02 10:47:20
昆明医科大学学报(2021年3期)2021-07-22 07:40:04
传染病信息(2021年6期)2021-02-12 01:52:58
生物学通报(2019年3期)2019-02-17 18:03:58
现代园艺(2017年13期)2018-01-19 02:28:09
现代检验医学杂志(2016年3期)2016-11-15 01:59:28
药学与临床研究(2015年4期)2015-06-05 11:35:54
生物医学工程学进展(2015年1期)2015-02-28 14:53:42
科学中国人(2015年16期)2015-02-28 09:14:02
