
microRNA-30 抑制血管紧张素Ⅱ诱导足细胞损伤的机制
2017-06-29 12:01张明超朱小东徐孝东施少林刘志红
肾脏病与透析肾移植杂志 2017年1期
赵 越 张明超 朱小东 徐孝东 杨 帆 郎 月 施少林 刘志红
microRNA-30 抑制血管紧张素Ⅱ诱导足细胞损伤的机制
赵 越 张明超 朱小东 徐孝东 杨 帆 郎 月 施少林 刘志红
目的:探讨microRNA-30(miR-30)缓解血管紧张素Ⅱ(AngⅡ)所致足细胞损伤的分子机制。 方法:(1)通过皮下埋置渗透压泵,给予小鼠AngⅡ[1 000 ng/(kg·min) ×28d]构建肾损伤模型。利用原位杂交及qRT-PCR技术检测小鼠肾小球中miR-30s的表达,运用免疫组化及蛋白印迹检测calcineurin信号重要分子细胞瞬时受体电位阳离子通道蛋白6(TRPC6),钙调磷酸酶(PPP3CA、PPP3CB、PPP3R1)及活化T细胞核因子( NFATC3)表达。(2)给予miR-30s慢病毒尾静脉注射,观察其对AngⅡ诱导损伤模型的肾脏保护作用。(3)体外研究观察miR-30a高表达能否抑制AngⅡ (10-6mol/L) 诱导的足细胞骨架损伤和足细胞凋亡。 结果:(1)AngⅡ诱导小鼠肾小球中miR-30s水平下调,并上调TRPC6,激活calcium/calcineurin信号通路;(2)高表达的miR-30s能够抑制AngⅡ导致的calcium/calcineurin信号活化从而缓解AngⅡ诱导的肾小球损伤;(3)miR-30s能够逆转AngⅡ所致足细胞骨架损伤及抑制其凋亡过程。 结论:miR-30s具有抑制AngⅡ诱导的足细胞损害作用,其机制可能与抑制calcium/calcineurin信号通路有关。
microRNA-30 血管紧张素Ⅱ calcium/calcineurin信号 足细胞
足细胞是一种终末分化的肾小球脏层上皮细胞,附着于肾小球基膜(GBM)表面,在维持肾小球滤过屏障完整性中起重要作用。足细胞损伤是导致蛋白尿、肾小球硬化及肾功能进行性恶化的重要病理基础[1]。因此,探讨足细胞损伤的分子机制将有助于寻找肾小球疾病的治疗靶点。
肾素-血管紧张素系统(RAS)的异常激活在多种原发和继发性肾脏疾病如局灶节段硬化性肾小球肾炎(FSGS)、糖尿病肾病及高血压导致的肾损伤的发生发展中起关键作用[2]。血管紧张素Ⅱ(AngⅡ)是RAS中的重要活性成分,可通过AngⅡ 1型受体(AT1R)调控肾脏疾病进展。有研究表明,足细胞过表达AT1R的转基因小鼠可出现足细胞损伤及凋亡的增加及肾小球的局灶节段硬化[3]。Ding等[4]证实AngⅡ可诱导大鼠足细胞骨架损伤,并有文献证实calcium/calcineurin信号通路的异常激活是AngⅡ所致足细胞凋亡及肾脏损伤的重要机制之一[5]。然而,AngⅡ通过何种机制激活calcium/calcineurin信号通路目前尚不明确。
微小核糖核酸(microRNA,简称miR)是一类长度约为18~25个核苷酸的内源性非编码小分子RNA,可抑制靶基因转录或促进目标mRNA降解,从而负性调节基因表达[6]。本课题组之前的研究证实miR-30家族(包括miR-30a、b、c、d、e五个成员)在肾小球内特异性表达于足细胞。给予体外培养的足细胞转化生长因子β(TGF-β)和嘌呤霉素核苷酸(PAN)等损伤因素刺激,可下调miR-30s家族成员表达水平[7]。疾病情况下调的miR-30s可直接作用于其靶基因TRPC6,PPP3CA,PPP3CB,PPP3R1和NFATC3,激活calcium/calcineurin信号通路,从而影响足细胞稳态及肾小球功能。基于calcium/calcineurin信号通路的异常激活可能是AngⅡ所致足细胞凋亡及肾脏损伤的重要分子机制,我们推测肾脏疾病情况下,AngⅡ很可能通过下调miR-30s,异常激活calcium/calcineurin通路,继而引发足细胞及肾小球损伤。本研究在前期研究的基础上,进一步通过体内研究完整阐释miR-30s在RAS系统调节肾脏生理病理过程中的重要作用,为更深入地了解肾小球疾病发生发展的机制,寻找新的足细胞损伤干预靶点提供理论依据。
材料和方法
实验动物 8周龄雄性C57BL/6J小鼠,体重20~25g,清洁级(南京军区南京总医院试验动物中心提供)共36只。
主要试剂 永生化的人足细胞株(HPC)由英国Bristol大学Moin. A. Saleem教授惠赠;RPMl l640培养基含10%胎牛血清、青霉素、链霉素各100 U/mL (美国Gibco)及1% insulin-transferrin-Selenium(Invitrogen公司)。其他试剂包括:Ang II (Sigma);TRPC6、PPP3CB和PPP3R1抗体(Abcam),PPP3CA抗体 (EMD Millipore),NFATC3抗体 (Santa Cruz,CA);RNA提取试剂盒(Ambion),TaqMan miRNA assay kit (ABI公司);Alexa Fluor@647 Annexin V和Propidium Iodide Solution (Biouniquer);罗丹明标记的鬼笔环肽(cytosleleton); microRNA原位杂交试剂盒(Exiqon)。采用Albuwell M 及 Creatinine companion kits (Exocell Laboratories)测定小鼠尿蛋白及尿肌酐。
实验方法
足细胞培养及处理 足细胞在33℃、5% CO2的培养箱中进行增殖,待细胞融合70%后转入37℃、5% CO2培养箱中分化10~14 d后,给予10-6mol/L AngⅡ刺激24 h,进行F-actin鬼笔环肽染色检测足细胞骨架损伤,Annexin V染色后结合流式细胞术检测细胞凋亡。
AngⅡ肾损伤模型构建 由于AngⅡ异常激活是慢性损伤的重要诱发因素,本研究造模采用8周龄C57BL/6J小鼠,给予皮下埋入胶囊渗透压泵(Alzet model 2004),恒量慢性输注AngⅡ[1 000 ng/(kg·min)]28d,以等量生理盐水作为对照。在AngⅡ输注后第14天,给予miR-30a慢病毒注射100 μl(浓度:1×108PFU/μl,汉恒生物)。28d后采用磁珠灌注法获取小鼠肾小球进行qPCR实验。
磁珠分选获取肾小球 小鼠麻醉后,给予含磁珠(Invitrogen,life technology)的PBS肾脏灌注,肾组织被切碎,消化30 min后过筛,用磁力架收集肾小球进行后续实验。
原位杂交实验 按照EXIQON-miRCURY LNATMmicroRNA ISH Optimization Kit (FFPE) 以及Roche- DIG Wash and Block Buffer Set试剂盒说明 进行小鼠肾脏组织原位杂交试验,检测miR-30a和miR-30d的表达及分布。
qRT-PCR检测 磁珠分选获取小鼠肾小球后,采用Ambion试剂盒提取总RNA,再使用逆转录试剂盒(TakaRa)进行cDNA合成后,采用TaqMan 探针法进行qPCR,检测 miR-30s的表达水平。
电镜检测 取小鼠肾组织,经3.75%戊二醛固定,2%锇酸固定,丙酮脱水、浸透,包埋、切片,染色,Hitachi 7500透射电镜观察。
Western Blot印迹 将组织用蛋白裂解液裂解后,加入上样缓冲液后煮沸变性。蛋白样品经电泳,转膜,封闭,一抗二抗孵育后,用增强的化学发光系统(ECL,Millipore)显色,在凝胶图像分析系统照相扫描蛋白条带的吸光度并分析处理。
免疫组化染色 取石蜡包埋的肾组织切片,经过二甲苯脱蜡、梯度乙醇复水,经微波修复10 min,10%小牛血清室温封闭10 min,一抗4℃孵育过夜,加入二抗,室温孵育30 min,进行DAB显色,封片后在显微镜下观察。
足细胞骨架损伤检测 接种于chamber slides(Nune)中的各组细胞,37℃ PBS洗三次,75%冰乙醇固定10 min后,洗去固定液,加入0.5% Triton X-100透膜5 min,洗涤,室温避光条件下鬼笔环肽孵育30 min,PBS洗三次,甘油封片,共聚焦荧光显微镜下(LSM510,Carl Zeiss)观察结果,扫描采集图像。
流式细胞仪检测细胞凋亡 收集各组细胞,按试剂盒(Biouniquer)方法,用Binding Buffer悬浮细胞后,在细胞悬浮液中加入Alexa Fluor 647 AnnexinV和PI,轻轻混匀后室温避光条件下孵育15 min,在1 h内用流式细胞仪(型号FACS ARIA,BD公司)检测细胞凋亡。
Tunel检测细胞凋亡 按照Tunel试剂盒In Situ Cell Death Detection Kit,POD (Roche)方法,选用小鼠肾组织石蜡切片,脱蜡后用0.1% Triton X-100通透细胞膜,PBS洗片后加入TUNEL混合液避光孵育1h,PBS漂洗三次,封片,荧光显微镜下观察,Image J软件进行计数。
统计学方法 应用SPSS 19.0统计软件进行分析,数据以均数±标准差表示,多组间比较采用单因素方差分析,两两比较用LSD检验,P<0.05为差异有统计学意义。
结 果
AngⅡ诱导小鼠肾损伤后足细胞中miR-30s的表达变化 我们将小鼠随机分为2组,分别给予AngⅡ和生理盐水慢性输注, 28d后采用磁珠灌注法获取小鼠肾小球进行qPCR实验,结果发现: AngⅡ能够下调miR-30家族成员水平(图1A)。小鼠肾组织切片原位杂交实验(ISH)结果提示:AngⅡ慢性处理28d后,小鼠足细胞中miR-30a和miR-30d水平均明显下调(图1B)。
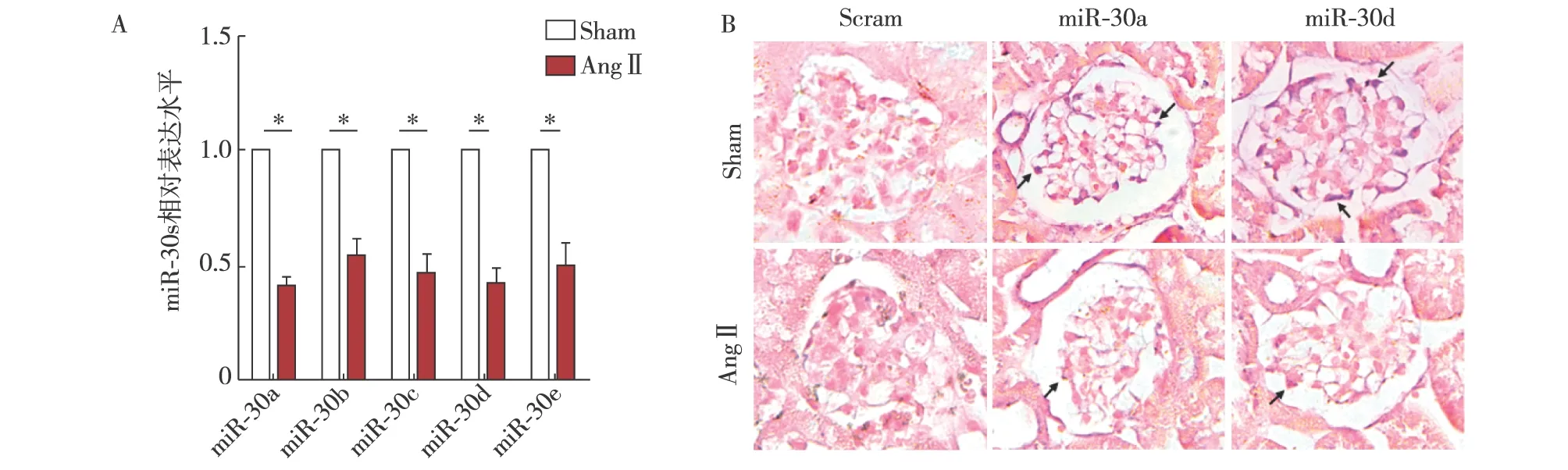
图1 AngⅡ处理可下调小鼠肾小球足细胞miR-30s水平miR-30:microRNA-30;Sham:生理盐水缓释组;AngⅡ:AngⅡ缓释组;Scram:AngⅡ小鼠给予Scramble慢病毒注射剂;A:qPCR分析显示AngⅡ慢性处理对小鼠肾小球中miR-30s表达水平的影响;B:原位杂交实验显示AngⅡ慢性输注对小鼠足细胞中miR-30a和miR-30d的影响;*:与生理盐水缓释组比较,P<0.05
高表达miR-30抑制AngⅡ所致小鼠肾脏损伤 给予AngⅡ慢性输注第一周,小鼠即可出现蛋白尿,一直持续到第四周(图2A)。在AngⅡ输注28d后,取肾组织进行PAS染色发现肾小球系膜扩张(图2B),电镜检测提示AngⅡ可诱导小鼠足细胞足突融合(图2C),进行Tunel实验检测足细胞凋亡情况,结果显示AngⅡ处理组小鼠足细胞凋亡较生理盐水处理组明显增加(图2D)。在AngⅡ慢性输注第14 天时,给予小鼠miR-30a慢病毒尾静脉注射, 28 天后处死小鼠,取其肾组织标本用作肾损伤评估,结果显提示miR-30a能够缓解AngⅡ所致的蛋白尿,肾小球系膜增生及足细胞损伤。

图2 miR-30抑制AngⅡ所致小鼠肾脏损伤miR-30:microRNA-30;Scram:给予 Scramble 慢病毒注射;miR-30a:给予miR-30a慢病毒注射;Vehicle:生理盐水缓释组;AngⅡ:血管紧张素Ⅱ;A:AngⅡ造模小鼠蛋白尿明显增加,miR-30a 治疗后可缓解其蛋白尿水平;B:AngⅡ造模28d后,小鼠肾脏病理切片PAS 染色显示肾小球系膜增生,而 miR-30a 治疗组肾损伤明显缓解;C:电镜显示AngⅡ造模小鼠出现足突融合, miR-30a 治疗组未出现该变化;D:Tunel实验提示AngⅡ慢性处理可诱导小鼠足细胞凋亡,而 miR-30a 可阻滞该作用;*:与Scram组比较,P<0.05;#:与AngⅡ+Scram组比较,P<0.05
miR-30s抑制AngⅡ诱导calcium/calcineurin信号活化 我们之前的研究已证实:calcium-calcineurin信号通路中的五个关键基因TRPC6,PPP3CA,PPP3CB,PPP3R1和NFATC3均为miR-30s的靶基因,并且通过体外实验证实miR-30s能够阻断AngⅡ所致calcium/calcineurin信号通路活化[8]。本研究进一步利用体内实验探讨miR-30s对AngⅡ所致calcium/calcineurin信号活化的影响。采用AngⅡ慢性输注28d的小鼠肾组织标本进行免疫组化染色及Western Blot实验,结果表明:AngⅡ能够上调TRPC6,PPP3CA,PPP3CB和PPP3R1,激活calcium/calcineurin通路,从而进一步活化NFATc3,给予miR-30慢病毒注射上调miR-30s表达后,AngⅡ诱导的calcineurin信号激活可被部分逆转(图3)。以上结果提示,AngⅡ可下调miR-30水平,进而激活calcineurin信号,导致足细胞损伤。
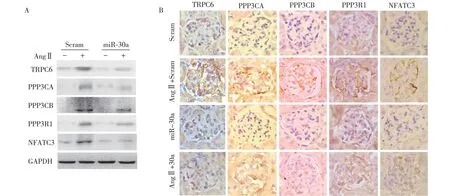
图3 miR-30s抑制AngⅡ诱导calcium-calcineurin信号活化miR-30s:microRNA-30s;TRPC6:细胞瞬时受体电位阳离子通道蛋白6;AngⅡ:血管紧张素Ⅱ;PPP3CA、PPP3CB、PPP3R1:钙调磷酸酶亚基;NFATC3:活化T细胞核因子;A: miR-30s对AngⅡ所致TRPC6、PPP3CA、PPP3CB、PPP3R1 及 NFATC 蛋白表达上调影响(Western Blot印迹);B:免疫组化检测上述蛋白在小鼠肾小球中的表达
miR-30s抑制AngⅡ所致体外培养的足细胞骨架损伤及凋亡 研究表明,AngⅡ能够诱导足细胞骨架损伤及凋亡[9]。体内外实验均证实,AngⅡ能够下调miR-30s水平,并且敲低miR-30能够导致足细胞骨架损伤和凋亡[7]。因此,我们推测miR-30s的下调参与了AngⅡ所致的足细胞损伤。为证明该假设,我们用10-6mol/L的AngⅡ处理体外培养的人永生化足细胞24h,进行F-actin鬼笔环肽染色及Annexin V染色。结果显示:AngⅡ刺激可导致足细胞F-actin压力纤维的丢失和足细胞凋亡率增加,而高表达miR-30s则能够部分逆转AngⅡ导致的足细胞损伤(图4),提示miR-30s介导了AngⅡ导致的足细胞骨架损伤及凋亡过程。
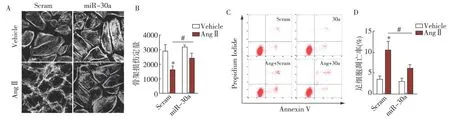
图4 miR-30a 可抑制 AngⅡ所诱导的足细胞损伤miR-30a:microRNA-30a;Scram:给予足细胞转染Scramble质粒;AngⅡ:血管紧张素Ⅱ;miR-30a:给予足细胞转染miR-30a质粒;Vehicle:空白对照组;A:荧光标记的鬼笔环肽显示转染Scram及miR-30a的足细胞给予AngⅡ处理后骨架损伤情况;B:A 图中骨架定量分析;C:通过流式细胞仪分析转染Scram及miR-30a的足细胞经过 AngⅡ处理 24h后 Annexin V 阳性细胞比例,从而反映足细胞凋亡情况; D:C 图中凋亡细胞定量;*:与Vehicle+Scram组比较,P<0.05;#:与AngⅡ+Scram组比较,P<0.05
讨 论
RAS异常活化在足细胞损伤、蛋白尿产生及肾小球硬化过程中发挥重要作用。AngⅡ 是 RAS中的重要活性成分,给予小鼠体内缓慢输注 AngⅡ 构建慢性肾损伤模型,可观察到小鼠出现蛋白尿,肾小球和肾小管损伤以及间质纤维化等病理改变。传统观点将 AngⅡ所致肾脏损伤机制归因于血流动力学作用,AngⅡ可增加肾小球毛细血管袢压力和滤过屏障通透性,进而诱发蛋白尿以及肾小管间质损伤[10]。近年研究提示 AngⅡ对肾脏细胞的直接生物学效应可能更为关键,而足细胞是其作用的主要靶细胞。大量研究表明,钙调磷酸酶(calcineurin)信号通路的异常活化是AngⅡ所致肾脏损伤及足细胞凋亡的重要机制之一。AngⅡ 可通过上调 TRPC6 通道蛋白表达水平,增加足细胞 Ca2+内流,活化calcineurin并刺激NFAT去磷酸化,同时上调细胞因子表达,刺激细胞外基质(ECM)产生,激活多种胞内信号途径导致细胞损伤[11-12]。 然而,AngⅡ 通过何种机制活化 calcineurin信号通路目前尚不明确。
多个团队研究证实,足细胞特异性 Dicer 酶敲除小鼠(该类小鼠足细胞成熟 miRNA 缺失)可出现显著的蛋白尿及肾小球硬化[13-14],提示miRNA 在维持足细胞的稳态过程中发挥不可或缺的作用。本课题组之前的研究发现:miR-30 家族在肾小球内特异性的表达于足细胞。且FSGS的患者及PAN诱导的足细胞损伤大鼠模型中,均可观察到miR-30家族成员的表达显著下降,给予PAN大鼠尾静脉 miR-30a 质粒注射后,足细胞及肾小球损伤可被逆转,并且蛋白尿水平明显缓解。体外实验中,我们给予人足细胞多种损伤因素刺激,如 TGF-β、脂多糖(LPS)及PAN后,miR-30s 的表达明显下调,而高表达 miR-30a 的人足细胞系可抵抗刺激因素所致损伤[7]。以上结果提示 miR-30s具有特异性的足细胞保护作用,进一步研究证实:miR-30s 的足细胞保护机制主要为靶向调控TRPC6、calcineurin(PPP3CA、PPP3CB、PPP3R1)以及 NFATC3[8]。从阻断钙内流,抑制 calcineurin 表达及 NFATC3 核转移三个层次紧密调控足细胞 calcineurin 信号通路。之前的研究中,我们已经通过体外实验证实,AngⅡ处理可显著下调足细胞miR-30s表达水平,上调calcium/calcineurin通路重要蛋白(TRPC6、PPP3CA、PPP3CB、PPP3R1以及NFATc3)表达。该结果提示,miR-30s在AngⅡ所致calcium/calcineurin信号通路异常激活中发挥重要作用。由于AngⅡ可作为循环激素在体内发挥重要调控作用,我们试图通过体内实验阐明miR-30s/calcineurin信号在RAS系统调节肾脏生理病理过程中的重要作用。我们给予小鼠皮下埋置胶囊缓释泵,利用AngⅡ慢性输注构建肾损伤模型,观察miR-30/calcineurin信号在AngⅡ所致足细胞损伤中的作用。AngⅡ慢性输注28d后即可观察到小鼠出现蛋白尿,系膜增生,足细胞骨架损伤等表型,提示AngⅡ所致慢性肾损伤模型构建成功。在此模型基础上,我们观察到TRPC6、calcineurin(PPP3CA、PPP3CB、PPP3R1)及 NFATC3表达上调而miR-30s表达被阻滞,提示AngⅡ处理可活化足细胞内calcineurin信号并下调miR-30家族表达。进一步给予小鼠尾静脉注射miR-30s慢病毒进行治疗,发现miR-30s能够显著缓解AngⅡ慢性输注所致蛋白尿及肾小球损伤表型。此外,miR-30s能够逆转AngⅡ所致TRPC6、calcineurin(PPP3CA、PPP3CB、PPP3R1)以及 NFATC3的表达上调。该结果表明:miR-30s能够逆转AngⅡ处理所致calcium/calcineurin信号通路的异常活化。
小结:本研究证实了AngⅡ通过下调miR-30s,进而活化calcium/calcineurin信号通路,最终导致足细胞损伤。我们的研究结果表明miR-30s下调不仅解释了AngⅡ活化calcium/calcineurin信号通路的分子机制,并且解读了AngⅡ导致足细胞损伤,包括足细胞骨架损伤和凋亡的原因。
1 Menon MC,Chuang PY,He CJ.The glomerular filtration barrier:components and crosstalk.Int J Nephrol,2012,2012:749010.
2 Kobori H,Nangaku M,Navar LG,et al.The intrarenal renin-angiotensin system:from physiology to the pathobiology of hypertension and kidney disease.Pharmacol Rev,2007,59(3):251-287.
3 Hoffmann S,Podlich D,Hähnel B,et al.Angiotensin II type 1 receptor overexpression in podocytes induces glomerulosclerosis in transgenic rats.J Am Soc Nephrol,2004,15(6):1475-1487.
4 Ding G,Reddy K,Kapasi AA,et al.Angiotensin II induces apoptosis in rat glomerular epithelial cells.Am J Physiol Renal Physiol,2002,283(1):F173-180.
5 Nijenhuis T,Sloan AJ,Hoenderop JG,et al.Angiotensin II contributes to podocyte injury by increasing TRPC6 expression via an NFAT-mediated positive feedback signaling pathway.Am J Pathol,2011,179(4):1719-1732.
6 Ambros V.The functions of animal microRNAs.Nature,2004,431(7006):350-355.
7 Wu J,Zheng C,Fan Y,et al.Downregulation of microRNA-30 facilitates podocyte injury and is prevented by glucocorticoids.J Am Soc Nephrol,2014,25(1):92-104.
8 Wu J,Zheng C,Wang X,et al.MicroRNA-30 family members regulate calcium/calcineurin signaling in podocytes.J Clin Invest,2015,125(11):4091-4106.
9 Jia J,Ding G,Zhu J,et al.Angiotensin II infusion induces nephrin expression changes and podocyte apoptosis.Am J Nephrol,2008,28(3):500-507.
10 Eckel J,Lavin PJ,Finch EA,et al.TRPC6 enhances angiotensin II-induced albuminuria.J Am Soc Nephrol,2011,22(3):526-535.
11 Wennmann DO,Hsu HH,Pavenstädt H.The renin-angiotensin-aldosterone system in podocytes.Semin Nephrol,2012,32(4):377-384.
12 Sonneveld R,van der Vlag J,Baltissen MP,et al.Glucose specifically regulates TRPC6 expression in the podocyte in an AngⅡ-dependent manner.Am J Pathol,2014,184(6):1715-1726.
13 Shi S,Yu L,Chiu C,et al.Podocyte-selective deletion of dicer induces proteinuria and glomerulosclerosis.J Am Soc Nephrol,2008,19(11):2159-2169.
14 Ho J,Ng KH,Rosen S,et al.Podocyte-specific loss of functional microRNAs leads to rapid glomerular and tubular injury.J Am Soc Nephrol,2008,19(11):2069-2075.
(本文编辑 青 松 加 则)
microRNA-30s reverse AngⅡ-induced podocyte injury
ZHAOYue,ZHANGMingchao,ZHUXiaodong,XUXiaodong,YANGFan,LANGYue,SHIShaolin,LIUZhihong
NationalClinicalReaearchCenterofKidneyDiseases,JinlingHospital,NanjingUniversitySchoolofMedicine,Nanjing210016,China
LIUZhihong(E-mail:liuzhihong@nju.edu.cn);SHIShaolin(E-mail:shaolinshil001@yahoo.com)
Objective:To illuminate the protective role of microRNA-30s in AngⅡ-induced podocyte injury. Methodology:(1) Mice were infused with AngⅡ at a dose of 1 000 ng/kg/min for 28 days with the osmotic mini-pumps implanted subcutaneously. The expression of miR-30s in mice glomeruli was examined by qRT-PCR and in situ hybrization. The expressions of TRPC6, PPP3CA、 PPP3CB、 PPP3R1 and NFATC3 were tested by western blotting and immunohistochemical. (2) To examine the protective effects of miR-30s in AngⅡ-treated mice, miR-30a-expressing lentivirus or control lentivirus was injected via the tail vein on day 14 during the AngⅡ infusion. The glomerular podocyte damage was evaluated with proteinuria,PAS staining and podocyte apoptosis and foot process effacement were used to measure podocye injury.(3) treated miR-30-overexpressed podocyte with 10-6mol/L AngⅡ, and cytoskeletal injury and cell apoptosis were examined by F-actin staing and Annexin V-flow cytometry. Results:(1) Ang II infusion induced the downregulation of miR-30 family and the activation of calcium/calcineurin signaling;(2)miR-30a could inhibit AngⅡ-induced activation of calcium/calcineurin signaling and then relieve AngⅡ-induced podocyte injury;(3) Exogenous miR-30a protected podocytes from AngⅡ-induced cytoskeletal damage and apoptosis in vitro. Conclusion:miR-30s could protect podocyte from AngⅡ-induced damage likely through calcium/calcineurin signaling inhibition.
microRNA-30s Angiotensin Ⅱ calcium/calcineurin signaling podocytes
10.3969/cndt.j.issn.1006-298X.2017.01.003
国际(地区)合作与交流项目(81320108007),国家自然科学基金青年基金(81600559),南京军区总院院管课题(2016035)
南京军区南京总医院肾脏科 国家肾脏疾病临床医学研究中心 全军肾脏病研究所(南京,210016)
刘志红(E-mail:liuzhihong@nju.edu.cn);施少林(E-mail:shaolinshil001@yahoo.com)
2016-11-25
ⓒ 2017年版权归《肾脏病与透析肾移植杂志》编辑部所有
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
昆明医科大学学报(2021年12期)2021-12-30
中国民间疗法(2021年19期)2021-11-20
中国民间疗法(2021年18期)2021-11-02
生物学通报(2019年3期)2019-06-15
中外医疗(2016年15期)2016-12-01
中国学术期刊文摘(2016年2期)2016-02-13
中国继续医学教育(2015年2期)2016-01-06
医学研究杂志(2015年9期)2015-07-01
河南医学研究(2014年1期)2014-02-27
